
ESR1基因及其功能
ESR1基因是雌激素受体(estrogen receptor,ER)基因,位于人类第6号染色体上,编码雌激素受体α蛋白(ERα)。ER是一种核受体,在体内负责介导雌激素的信号,调节生殖系统的生长、分化和各种生理功能。该基因突变可能会导致雌激素信号通路的异常,从而对肿瘤细胞的生长和转移产生影响。
ESR1基因突变及其影响
ESR1突变在乳腺癌患者中的发生率取决于既往内分泌治疗场景及治疗持续时间,在HR阳性乳腺癌中,ESR1突变大多发生于转移性疾病患者芳香化酶抑制(AI)治疗后 1。大约20%~40%的转移性乳腺癌(MBC)患者AI治疗后出现ESR1突变,其发生率因转移性病灶位置而异。相反,AI辅助治疗或新辅助治疗后复发的乳腺癌患者中ESR1突变率分别为4~5%和1.5~7%,而未接受过内分泌治疗的MBC患者ESR1突变率小于1% 1。
ESR1突变的常见类型包括点突变、插入/缺失和重排等。ESR1突变通常与多个基因组改变同时发生,共突变往往导致总体预后较差 1。所有ESR1耐药突变均位于配体结合结构(ligand-binding domain,LBD)结构域 (图1),最常见的突变类型是D538G和Y537S,其他包括Y537N、Y537C、L536H、L536P、L536R、S463P、E380Q1-2。大多数ESR1突变位于ER LBD关键螺旋12区域内或附近的少数氨基酸,主要以单等位基因突变形式存在(图1)2。LBD突变型ESR1(以下简称ESR1突变)具有稳定的活性构象,增加了与共刺激因子的结合,并减少了ERα蛋白水解降解 (图2)。ESR1基因融合虽然较为罕见,但因融合蛋白缺失LBD,其表达导致配体不依赖的肿瘤生长,及对选择性雌激素受体调节剂(SERM)类药物和氟维司群的完全耐药3。大约50%的内分泌治疗耐药与ESR1突变相关 1。另外,ESR1突变还可以影响ERα与其他信号通路蛋白的相互作用,如PI3K/AKT和MAPK等信号通路,从而影响肿瘤细胞的增殖和转移能力。
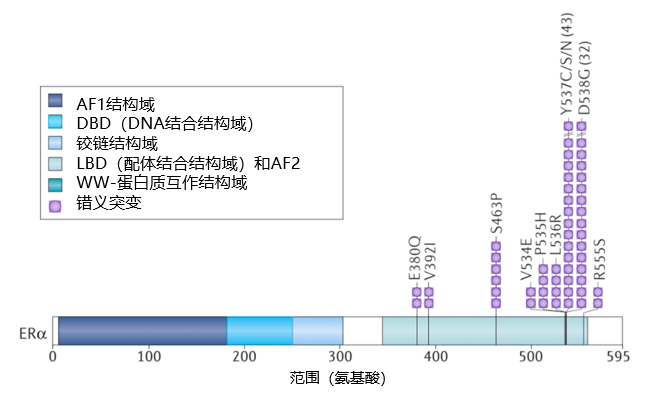
图1 ESR基因和最常见突变 2
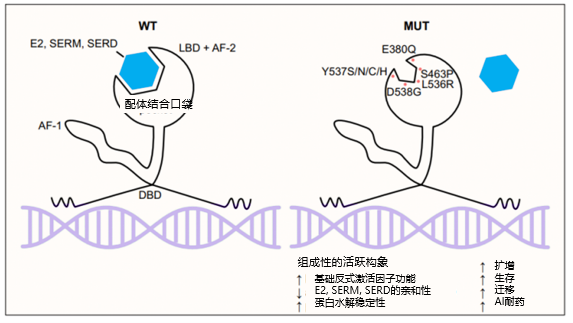
图2 LBD结构域中的所有热点ESR1突变及其影响 1
ESR1突变在乳腺癌中具有重要的临床意义 1,3,4。一方面,ESR1突变是激素治疗耐药的重要机制之一。药代动力学显示,他莫昔芬与ESR1突变体的结合亲和力下降达30倍,而氟维司群与ESR1突变体的结合亲和力下降40倍,因此可能需要更高的剂量水平来抑制其功能。另外不同的突变形式的临床意义和对药物的响应情况可能也不尽相同(图3)。例如,在两个最常见的突变中,Y537S与D538G相比,对他莫昔芬、氟维司群和新型SERM或SERD类药物具有更大的耐药性。另一方面,D538G变异的转移潜力更大,特别是转移到肝脏,而且与Y537S不同,可能会增加Wnt信号通路的转导。因此,对于转移性乳腺癌患者,检测ESR1突变状态有助于选择更合适的治疗方案。
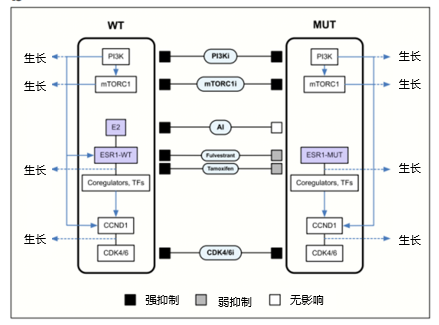
图3 HR阳性乳腺癌中的关键靶向通路及ESR1突变的影响
另一方面,ESR1突变体也可以作为治疗靶点。例如,对于Y537S突变患者,目前已经开发出针对该突变的靶向药物。但是,ESR1突变的种类和数量非常复杂,针对不同突变类型的靶向药物还需要进一步的研究和验证。此外,ESR1突变还与其他预后指标相关,如较短的无进展生存期和较差的总生存率。因此,ESR1突变检测也可以用于评估患者的预后风险和制定个体化治疗策略。
ESR1突变患者的内分泌治疗选择
ESR1突变可以通过小分子靶向药阻滞其下游通路产物进行抑制(如PI3K抑制剂,mTOR抑制剂及CDK4/6抑制剂,图3),尽管ESR1突变与AI治疗的不良预后相关,其他内分泌治疗(ET)方式仍可以作为选择。
氟维司群是一种选择性雌激素受体拮抗剂(Selective Estrogen Receptor Downregulator, SERD),通过与ER结合并诱导受体降解来抑制雌激素依赖性肿瘤的生长5。临床前研究显示ESR1突变体需要更高剂量的氟维司群来发挥疗效,然而临床研究中相同剂量水平下的ESR1野生型及突变型晚期乳腺癌患者的PFS相似6,7,单纯的ESR1突变可能并不能带来对氟维司群的耐药。然而,由于存在多种ESR1突变以及个体差异,氟维司群对所有ESR1突变型乳腺癌患者的疗效可能有所不同。因此,针对ESR1突变型乳腺癌的最佳治疗策略需综合患者具体情况个性化制定治疗选择。
口服SERD类药物
目前指南推荐,转移性乳腺癌患者在接受联合化疗前接受2-3线的内分泌治疗。对于ET耐药风险较高的患者,如ESR1突变型转移性乳腺癌患者的AI耐药,需要额外的ET选择。对几种新型SERM和SERD评估的临床试验正在进行中。口服类SERD药物是一种新型的内分泌治疗药物,可以直接口服而不需要注射。与传统注射给药相比,口服用药具有更好的吸收效果和更持久的作用时间。口服药物可以在胃肠道中被吸收,并通过肝脏代谢后进入血液循环。由于口服给药不受第一次通道代谢的影响,因此其生物利用度更高,可以在保证疗效的同时有效降低药物剂量以减少副作用。不同SERD与ER的相互作用存在差异,可根据结构特征和作用机制对口服SERD类药物进行分类:
- 具有ER结合位点和丙烯酸侧链的非甾体分子。
- 具有氨基侧链,诱导ER发生不同的构象变化,影响转录活性并使ER易于降解
口服类SERD药物在ER阳性晚期乳腺癌患者中已被证明是一种有效的治疗方法,尤其是在接受过其他内分泌治疗且治疗失败的患者中。在晚期乳腺癌中(尤其是ESR1突变患者),无论是作为CDK4/6抑制剂进展后单药治疗,还是与CDK4/6抑制剂联合一线治疗,口服SERD均表现出了可观的临床获益。而在早期乳腺癌中,口服SERD由于其低毒性,或许有助于提高辅助及新辅助治疗患者的依从性及耐受性。
靶向ER的PROTAC类药物
PROTAC(Proteolysis Targeting Chimeras,蛋白酶解靶向嵌合物)是一类可特异性诱导蛋白质降解的小分子化合物。其作用机制是通过诱导细胞内的蛋白降解系统,将特定的病理性蛋白降解,从而达到治疗目的。PROTAC分子通常由三部分组成:1)靶向蛋白的小分子配体;2)泛素连接酶(E3 ligase)的小分子配体;3)连接配体的链接子。在细胞内,PROTAC分子将靶蛋白与E3连接酶聚集在一起,形成一个底物-泛素连接酶-PROTAC复合物,促使靶蛋白被泛素化。泛素化的靶蛋白随后被蛋白酶体降解系统识别和降解。
研究显示PROTAC药物可有效降解野生型和突变型(包括ESR1突变型)雌激素受体。与氟维司群相比,PROTAC类药物在雌激素依赖性乳腺癌中的的抑制作用更强。此外,PROTAC药物在乳腺癌动物模型中显示出良好的抗肿瘤活性,可显著缩小肿瘤体积。因此,PROTAC药物可能为乳腺癌患者提供一种有效的治疗策略,特别是在携带ESR1突变和内分泌治疗耐药的患者中。
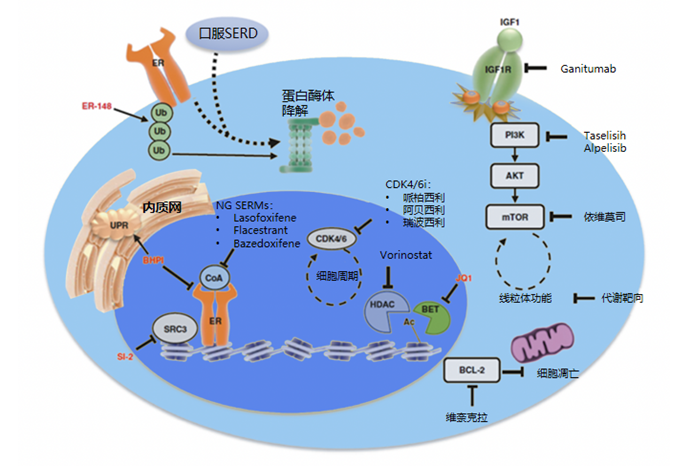
图4 ESR1突变乳腺癌潜在治疗靶点
结论
自1997年发现MBC中的ESR1突变以来,改进的临床前模型和对大量患者样本的基因组分析已将这些突变与ET耐药性严重联系起来。在AI治疗的MBC中,有明显的获得性ESR1突变的且影响其对AI疗效。尽管在氟维司群经治的患者中ESR1 Y537S突变率有所增加,但SERDs在ESR1突变的MBC中的作用仍不清楚。目前也有很多新型内分泌治疗药物的研究在进行中,并确定对其ESR1突变体临床前模型中的有效性,以改善ESR1突变体MBC患者的预后。目前早期研究显示新型内分泌治疗药物,如口服SERD类药物及PROTAC类药物等,为ESR1突变患者提供了有效的治疗选择。此外,相较于传统SERD类药物,PROTAC药物具有更高的靶蛋白降解率,能够更有效地抑制ESR1突变乳腺癌。
参考文献
- Brett JO, et al. Breast Cancer Res. 2021 Aug 15;23(1):85.
- Reinert T, et al. Front Oncol. 2017 Mar 15;7:26.
- Jeselsohn R, et al. Nat Rev Clin Oncol. 2015 Oct;12(10):573-83.
- Herzog SK, et al. Br J Cancer. 2022 Feb;126(2):174-186.
- Carlson RW. Clin Breast Cancer. 2005 Apr;6 Suppl 1:S5-8.
- Turner NC, et al. Clin Cancer Res. 2020;26:5172–7.
- Clatot F, et al. Oncotarget. 2016;7:74448–59.
文档编号:PP-UNP-CHN-0396


