
细胞周期调控机制的失调,尤其是细胞周期蛋白依赖性激酶4和6的过度激活,是乳腺癌发病的重要特征。CDK4/6抑制剂的激活通过靶向异常细胞周期进程,彻底改变了激素受体阳性乳腺癌的治疗格局。然而,尽管这类药物初期取得临床成功,但耐药性仍是主要挑战,目前尚缺乏可靠的生物标志物来预测治疗反应或指导耐药人群的管理策略。因此,众多研究致力于探索耐药机制,以优化CDK4/6抑制剂的临床应用并改善患者预后。本文系统阐述了细胞周期的分子调控机制、CDK4/6抑制剂在乳腺癌中的临床应用现状,以及耐药形成的关键机制。此外,还探讨了新兴预测性生物标志物,并提出了克服耐药性和提升疗效的潜在发展方向。
一、细胞周期调控机制
细胞周期的顺利进展需要严格的调控,特别是在G1期至S期的过渡中,视网膜母细胞瘤蛋白和E2F转录因子之间的相互作用起到了决定性作用。在正常的细胞周期过程中,有丝分裂信号刺激可诱导细胞周期蛋白D的表达,从而结合并激活CDK4/6,形成具有活性的cyclin D–CDK4/6复合物。激活后的CDK4/6通过磷酸化视网膜母细胞瘤蛋白,促使E2F转录因子有效释放。被释放的E2F启动参与S期进入的相关基因转录,包括CDK2激活因子细胞周期蛋白E和细胞周期蛋白A。CDK2的激活会进一步磷酸化视网膜母细胞瘤蛋白及DNA合成与复制所需的各种底物,从而驱动细胞进入S期。然而,当这一过程受到干扰,尤其是cyclin D或CDK4/6的过度激活时,细胞就会进入不受控制的增殖状态,形成肿瘤。
总而言之,细胞周期包含一系列严格调控的阶段,确保细胞能够有序完成生长、DNA复制与分裂过程。该过程的关键调控因子包括细胞周期蛋白、细胞周期蛋白依赖性激酶、CDK抑制蛋白以及视网膜母细胞瘤蛋白,这些因子共同调控不同细胞周期阶段之间的转换。这些调控分子的改变可能导致肿瘤细胞中异常增殖现象的发生。在50%-60%的乳腺癌病例中可观察到cyclin D过表达,这提示CDK4/6存在过度激活。此外,cyclin E过表达、CDK4/6基因扩增以及p53等抑癌基因的功能缺失突变也常见于乳腺癌,这些变异共同诱导细胞周期异常进展。这些对细胞周期启动经典与非经典通路的全面理解,揭示了细胞增殖中复杂的调控网络,为治疗调控机制失调相关癌症提供了潜在的靶点和策略(图1)。
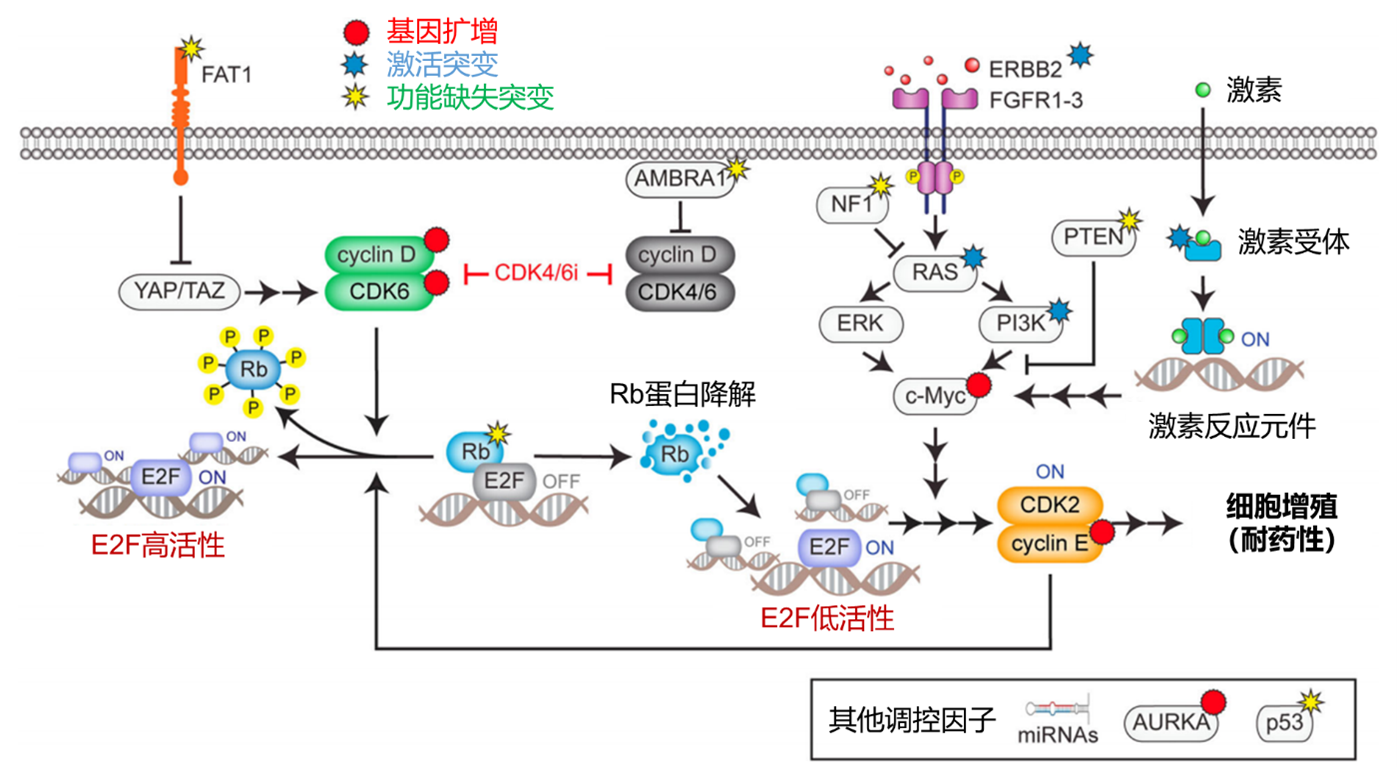
图1. CDK4/6抑制剂耐药机制
该示意图展示了激素受体阳性/人表皮生长因子受体2阴性乳腺癌中对CDK4/6抑制剂产生耐药的关键遗传与非遗传机制。在经典通路中,有丝分裂信号与激素信号通路激活细胞周期蛋白D-CDK4/6复合物,该复合物通过磷酸化视网膜母细胞瘤蛋白释放E2F转录因子,进而促进细胞周期启动。视网膜母细胞瘤蛋白基因功能丧失突变是最常见的CDK4/6抑制剂原发性耐药机制。另一种遗传机制涉及FAT1功能缺失突变,该突变通过激活Hippo通路导致CDK6过表达,并形成CDK6‒INK4复合物,从而使肿瘤细胞对CDK4/6抑制剂产生耐药性。在耐药肿瘤中常观察到有丝分裂信号与激素信号通路相关基因(如ERbB2、FGFR1-3、PIK3CA、NF1和ESR1)突变,这些突变可上调c-Myc表达。非遗传机制包括视网膜母细胞瘤蛋白降解,通过绕过CDK4/6抑制作用以其他方式进入细胞周期。然而这种非经典视网膜母细胞瘤蛋白失活途径并不完整,需要c-Myc对E2F进行扩增,已有研究表明该机制与CDK4/6抑制剂疗效呈负相关。AMBRA1缺失可稳定cyclin D并促进其与CDK2结合,从而引发耐药。此外,AURKA和CCNE1/2扩增可上调CDK2活性,使肿瘤细胞摆脱对CDK4/6的依赖。特定微小 RNA的改变也会导致 CDK4/6 抑制剂耐药。该模型强调了耐药机制的多维性,揭示了基因突变、信号通路异常和转录调控在CDK4/6 抑制剂耐药中的重要作用。
二、CDK4/6抑制剂在乳腺癌治疗中的作用
CDK4/6抑制剂通过阻断细胞周期在G1期的进程并诱导细胞衰老发挥作用。HR+/HER2−乳腺癌通常保留视网膜母细胞瘤蛋白功能,并通过雌激素信号激活cyclin D-CDK4/6通路,因此对CDK4/6抑制的敏感性最高。这类抑制剂显著的疗效推动了一系列临床试验的开展,随后美国食品药品监督管理局批准了CDK4/6抑制剂联合内分泌疗法用于HR+/HER2−转移性乳腺癌。
三、CDK4/6抑制剂的耐药机制及疗效预测的潜在生物标志物
3.1. 遗传性耐药机制
在HR+/HER2−乳腺癌患者中,约30%对CDK4/6抑制剂产生耐药性的患者会出现新的基因突变。由于视网膜母细胞瘤蛋白是CDK4/6的主要作用底物,视网膜母细胞瘤蛋白基因LOF突变成为临床前研究中证据最充分的耐药机制。携带视网膜母细胞瘤蛋白基因LOF突变的乳腺肿瘤细胞对CDK4/6抑制剂无应答,但通过重新引入野生型视网膜母细胞瘤蛋白可逆转这种原发性耐药。然而在HR+/HER2−乳腺癌中,视网膜母细胞瘤蛋白 LOF突变相对罕见,仅见于4.7%的病例。另一种值得关注的突变是FAT1基因LOF突变,在HR+/HER2−乳腺癌中约有6%的患者存在FAT1基因LOF突变。此类突变通过Hippo信号通路驱动CDK6过表达,进而形成对CDK4/6抑制剂耐药的CDK6–INK4复合物。此外,CDK6基因扩增及特异性CDK6复合物的形成也被多项研究证实与耐药机制相关。
有丝分裂信号通路和激素信号通路可诱导cyclin D表达,从而激活CDK4/6,这一过程对CDK4/6抑制剂的敏感性具有决定性意义。然而大量研究证实,有丝分裂信号通路和激素信号通路的异常激活与CDK4/6抑制剂耐药相关。例如,耐药肿瘤中常见FGFR信号通路扩增和PTEN基因缺失,而KRAS基因突变则与HR+/HER2−乳腺癌患者更短的PFS相关。大多数导致耐药的驱动突变存在于有丝分裂和激素信号相关基因中(如PIK3CA、ESR1、ERbB2、NF1及FGFR1等)。值得注意的是,无论ESR1基因是否突变,乳腺癌仍可能对CDK4/6抑制剂产生应答;且PIK3CA突变在敏感型与耐药型肿瘤中均存在。这些发现提示,上述基因变异可能在维持初始治疗敏感性的同时,逐步介导耐药机制的形成。
3.2. 非遗传性耐药机制
约 70% 初始对 CDK4/6 抑制剂产生应答的 HR+/HER2−乳腺癌患者,在未伴随新的体细胞突变的情况下会产生耐药性,这提示非遗传机制的重要作用。既往研究揭示,HR+乳腺肿瘤细胞中会形成非典型cyclin D–CDK2复合物,通过替代性激活CDK2导致耐药。AMBRA1蛋白缺失可促进cyclin D过表达,进而增强其与CDK2的相互作用。最新研究还发现,非磷酸化视网膜母细胞瘤蛋白本身不稳定,易发生蛋白酶体降解。虽然这种视网膜母细胞瘤蛋白降解能不依赖CDK4/6启动E2F激活,但仅能实现E2F的部分低水平激活。全局转录放大器c-Myc通过增强这种低水平E2F活性,在CDK4/6抑制剂耐药发展中起关键作用。因此,在治疗前样本中,c-Myc的表达水平与HR+/HER2−乳腺癌一线CDK4/6抑制剂联合内分泌治疗的疗效呈负相关。
调控G2/M期转换的极光激酶A基因扩增现象,在对CDK4/6抑制剂耐药的HR+乳腺癌中发生率达27%。相比之下,对CDK4/6抑制剂敏感的肿瘤则保持正常的AURKA表达水平。细胞周期蛋白 E1和 E2的扩增也常与耐药性相关,细胞周期蛋白E的上调可增强CDK2活性,从而绕过对CDK4/6通路的依赖。一项研究发现,在对CDK4/6抑制剂产生获得性耐药的HR+/HER2−细胞系中存在CCNE1扩增。抑制CCNE1或CDK2能够恢复细胞对CDK4/6抑制剂的敏感性,这证实了CCNE1在耐药细胞中驱动CDK2依赖性细胞周期进程的关键作用。
非编码RNA分子 microRNA 亦被证实参与介导对CDK4/6抑制剂的耐药性。例如,在耐药的HR+/HER2−细胞中,miRNA-432-5p的上调会抑制转化生长因子 β通路中的SMAD4,进而导致CDK6表达上调并引发 对CDK4/6 抑制剂耐药。类似地,miRNA-29b-3p也能调控CDK6的表达及耐药性。此外,miRNA-223被认为是潜在的耐药标志物,其表达缺失与乳腺癌患者总生存期降低相关。其他miRNA(包括miRNA-126、miRNA-326和miRNA-3613-3p)在乳腺肿瘤细胞系中也显示出与CDK4/6抑制剂的敏感性相关。
3.3 CDK4/6抑制剂疗效的潜在预测性生物标志物
在HR+/HER2-乳腺癌患者中,CDK4/6抑制剂的治疗效果存在个体差异,这凸显了寻找能预测疗效的可靠生物标志物的迫切需求。关于CDK4/6抑制剂疗效的潜在预测性生物标志物总结如表1所示。
表1. CDK4/6抑制剂疗效的潜在预测性生物标志物
生物标志物类别 | 临床/证据说明 | 备注 |
|---|---|---|
视网膜母细胞瘤蛋白基因LOF突变 | 最显著的预测生物标志物之一;在胚系BRCA2突变乳腺癌中富集,导致PFS差;然而在HR+/HER2−乳腺癌中,视网膜母细胞瘤蛋白基因LOF突变在临床上较为罕见,仅见于约4.7%的病例。 | 原发性耐药的明确驱动因素 |
RbmRNA/蛋白水平 | 在视网膜母细胞瘤蛋白功能完整的患者中,其表达水平变化不能可靠预测治疗反应。 | 无可靠预测价值 |
Cyclin D mRNA/蛋白水平 | 临床数据表明基线Cyclin D水平与患者对CDK4/6抑制剂的反应无显著相关性;Cyclin D与Cip/Kip家族成员的比值,比单独的Cyclin D水平更能可靠地预测CDK4/6的激活状态。 | 无可靠预测价值 |
FAT1基因LOF突变 | 对HR+/HER2−乳腺癌基线活检样本的基因组学分析显示,FAT1基因LOF突变是预测CDK4/6抑制剂治疗早期进展的重要指标。这些突变约见于6%的转移性样本和2%的原发肿瘤样本。 | 潜在预测性生物标志物 |
TP53基因LOF突变 | 虽然TP53基因LOF突变与肿瘤更具侵袭性和OS缩短相关,但它并不必然导致对CDK4/6抑制剂耐药。相反,TP53突变肿瘤更可能在CDK4/6抑制剂治疗中表现出快速进展。 | 预后不良的影响因素 |
有丝分裂/激素信号通路与c-Myc | 对HR+/HER2−乳腺癌样本的基线分析显示,提示PI3K通路激活的磷酸化AKT,以及c-Myc基因扩增和c-Myc蛋白水平升高,与接受CDK4/6抑制剂联合内分泌治疗患者的较差PFS相关。这些发现表明,c-Myc及其调控通路可作为预测乳腺癌CDK4/6抑制剂疗效的生物标志物。 | 潜在预测性生物标志物 |
CCNE1扩增 | CCNE1扩增是一种已知的CDK4/6抑制剂耐药机制。在乳腺癌活检组织中,CCNE1过表达与对CDK4/6抑制剂的耐药显著相关,而HR+乳腺癌中CCNE1 mRNA水平较低则与更好的治疗应答相关。 | 耐药机制与潜在预测性生物标志物 |
四、克服CDK4/6抑制剂耐药性的潜在策略
为了克服耐药性,研究者提出了将CDK4/6抑制剂与其他靶向药物联合使用的治疗策略。特别是针对PI3K/AKT/mTOR信号通路的抑制,可以在CDK4/6抑制剂耐药的情况下提供新的治疗思路。例如,在CDK4/6抑制剂治疗后出现疾病进展且携带PIK3CA突变的乳腺癌患者,联合使用PI3K抑制剂与内分泌治疗可以显著改善患者的预后。此外,多项临床试验对CDK4/6抑制剂耐药后进行CDK4/6抑制剂联合内分泌维持治疗的潜在价值进行了探索。临床研究表明,在接受CDK4/6抑制剂治疗耐药后换用另一种CDK4/6抑制剂可能带来获益。近期一项临床前研究揭示,在耐药细胞中持续进行CDK4/6抑制会激活一条非经典通路,导致视网膜母细胞瘤蛋白发生降解(而非通过磷酸化失活)。这一机制引发的E2F激活动力学缓慢且水平较低,从而显著减弱了耐药肿瘤的生长。值得注意的是,这种被削弱的E2F活性虽能减缓细胞周期进程,却不影响细胞进入周期。这些结果凸显了二线治疗方案应优先选择维持靶向治疗,而非转向全身性化疗的重要性。
总结
靶向乳腺肿瘤细胞周期调控机制是提升治疗效果的潜在策略。尽管CDK4/6抑制剂已重塑治疗格局,但克服耐药性、探索新靶点与创新疗法对维持其疗效至关重要。未来研究应聚焦联合治疗方案、非经典细胞周期启动通路及肿瘤微环境,或将为开发更有效持久的治疗方案奠定基础。将这些策略整合至临床实践中,最终将改善乳腺癌患者的疾病管理与预后。
参考文献
- Shanabag A, Armand J, Son E, Yang HW. Targeting CDK4/6 in breast cancer[J]. Exp Mol Med. 2025 Feb;57(2):312-322.



