请仔细阅读说明书并在医师指导下使用
本品为注射用舒巴坦钠与注射用度洛巴坦钠的组合包装。
1、注射用舒巴坦钠
本品活性成份为舒巴坦钠。
化学名称:(2S,5R)-3,3-二甲基-7-氧代-4-硫杂-1-氮杂双环[3.2.0]庚烷-2-羧酸钠- 4,4-二氧化物
化学结构式:
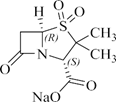
分子式:C8H10NNaO5S
分子量:255.22
辅料:本品不含辅料。
2、注射用度洛巴坦钠
本品活性成份为度洛巴坦钠。
化学名称:(2S,5R)-2-氨甲酰基-3-甲基-7-氧代-1,6-二氮杂双环[3.2.1]辛-3-烯-6-基硫酸钠
化学结构式:
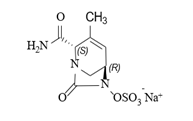
分子式:C8H10N3NaO6S
分子量:299.23
辅料:氢氧化钠、盐酸。
本品用于治疗 18 岁及以上患者由鲍曼-醋酸钙不动杆菌复合体敏感分离株所致医院获得性细菌性肺炎(HABP)、呼吸机相关性细菌性肺炎(VABP)。
本品不适用于治疗鲍曼- 醋酸钙不动杆菌复合体敏感分离株以外病原体所致HABP/VABP(见【临床试验】)。
为减少耐药菌形成,维持本品和其它抗菌药物的有效性,本品仅用于治疗已证实或强烈疑似由敏感细菌引起的感染。当可获得培养及药敏信息,选择或调整抗菌治疗时应予以考虑。如缺少此类数据,根据经验进行治疗选择时可参考当地流行病学和药敏性特征信息。
每个组合包装含注射用舒巴坦钠(1.0g,按 C8H11NO5S 计)1 瓶与注射用度洛巴坦钠(0.5g,按C8H11N3O6S 计)2 瓶。
推荐剂量
本品是含注射用舒巴坦钠和注射用度洛巴坦钠的组合包装产品。肌酐清除率(CLcr)为 45-129 mL/min 的成人患者,本品推荐剂量为 1.0 g 舒巴坦和 1.0 g 度洛巴坦,每 6 小时进行一次静脉(IV)输注,每次持续 3 小时。
对于 CLcr<45 mL/min 和 CLcr≥130 mL/min 的患者,建议调整本品给药方案。
建议本品治疗持续时间为 7-14 天。应根据患者的临床状态,选择治疗持续时间。
18 岁及以上患者根据肾功能的用药剂量推荐
18 岁及以上患者根据肾功能的用药剂量推荐见表 1。
对于 CLcr<45 mL/min 和肾功能亢进(CLcr≥130 mL/min)的患者,建议调整给药方案。对于接受间歇性血液透析(HD)患者,在 HD 完成后立即开始本品给药。
对于肾功能波动患者,应监测 CLcr 并相应调整剂量。
表1:18 岁及以上患者根据肾功能的用药剂量推荐
舒巴坦和度洛巴坦的剂量(g) | 估算的 CLcr | 给药频率 |
|---|---|---|
| 舒巴坦 1.0g 和度洛巴坦 1.0g | ≥ 130 | 每 4 小时 1 次 |
| 45-129 | 每 6 小时 1 次 | |
| 30-44 | 每 8 小时 1 次 | |
| 15-29 | 每 12 小时 1 次 | |
| < 15† | 对于开始使用本品的患者:前 3 次给药每 12 小时 1 次(0、12 和 24 小时),第 3 次给药后每 24小时 1 次† 对于目前正在接受本品且 CLcr 下降至 15mL/min 以下的患者:每 24 小时 1 次 |
*CLcr=用 Cockcroft-Gault 方程估算的肌酐清除率
†对于接受血液透析的患者,应在透析疗程结束后给药
接受连续性肾脏替代治疗(CRRT)的患者
在这种情况下,提供剂量调整的信息有限,应根据患者的临床状态指导治疗。在 CRRT 期间,患者的残余肾功能可能发生变化,可能需要调整本品剂量。治疗过程中肾功能可能发生变化,需定期监测肾功能,并相应调整本品剂量。
肝功能损害
尚未评估肝功能损害对舒巴坦和度洛巴坦药代动力学的影响。因舒巴坦和度洛巴坦均未发生实质性肝脏代谢/排泄,预计肝功能损害不会影响本品消除。肝功能损害患者无需调整剂量。
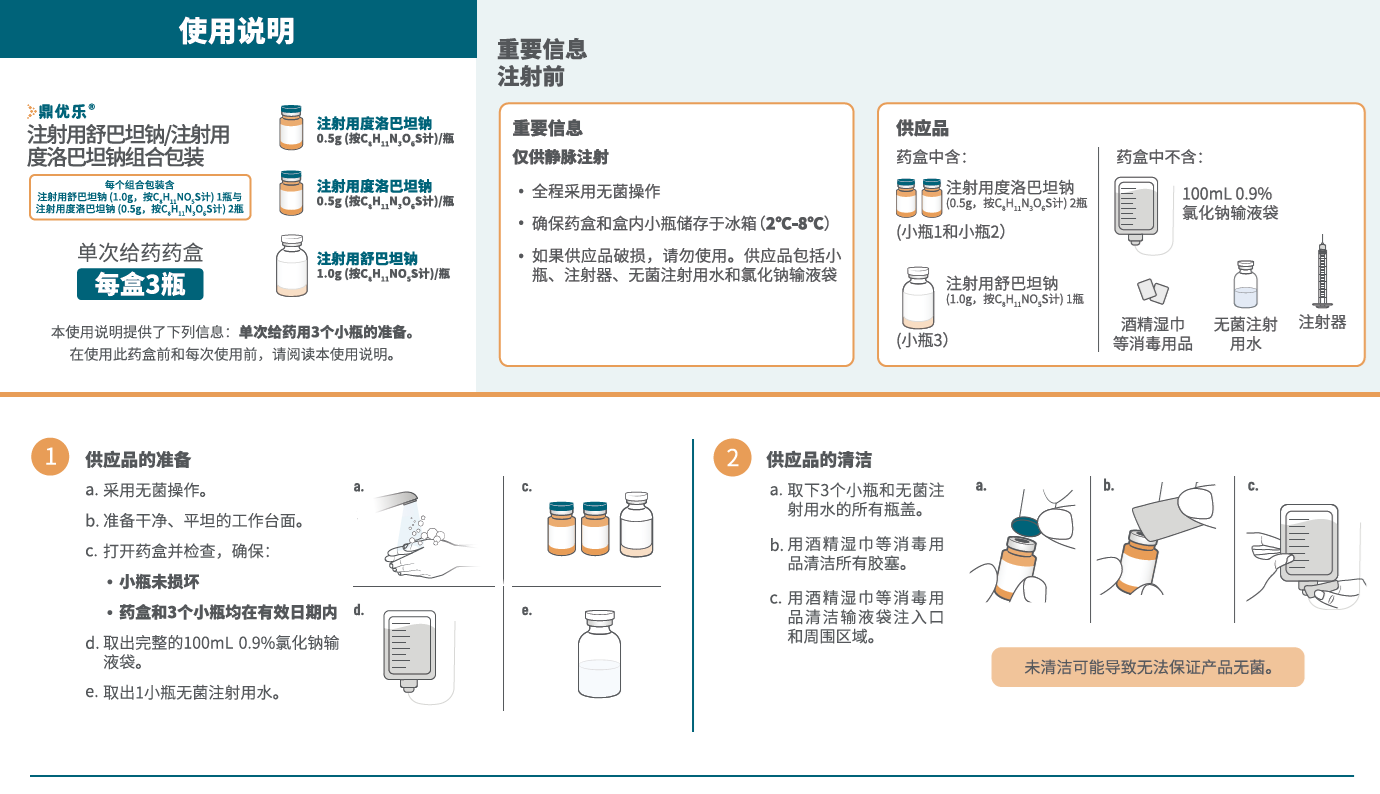
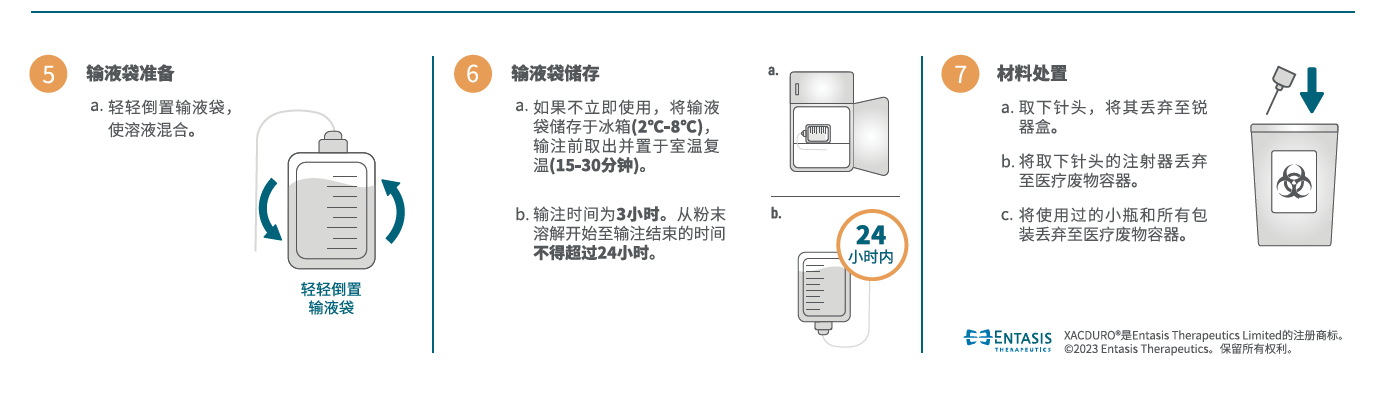
本品静脉给药用溶液的制备、储存和给药方法
本品为一种组合包装药盒,含 1.0 g 舒巴坦(无菌粉末)的一个透明单剂量小瓶和每瓶含 0.5 g 度洛巴坦(无菌粉末)的两个棕色单剂量小瓶,静脉输注前须使用无菌操 作进行复溶和进一步稀释。本品不含抑菌防腐剂,已稀释的输注溶液,如果不立即使用,需要在2-8℃的冷藏条件下储存(不得冷冻),并在使用前置于室温下复温(15-30分钟)。每次静脉输注时间持续3小时,且从粉末溶解开始至输注结束的时间不得超过24小时。未用完药液应丢弃。
本品制备所需项:
- 本品组合包装(包括1个每瓶含 1.0 g 舒巴坦的单剂量小瓶和 2 个每瓶含0.5 g 度洛巴坦的单剂量小瓶)
- 含100 mL 0.9% 氯化钠注射液的输液袋
- 10 mL 无菌注射用水
- 10 mL 无菌注射器和酒精棉等消毒用品
本品的制备:
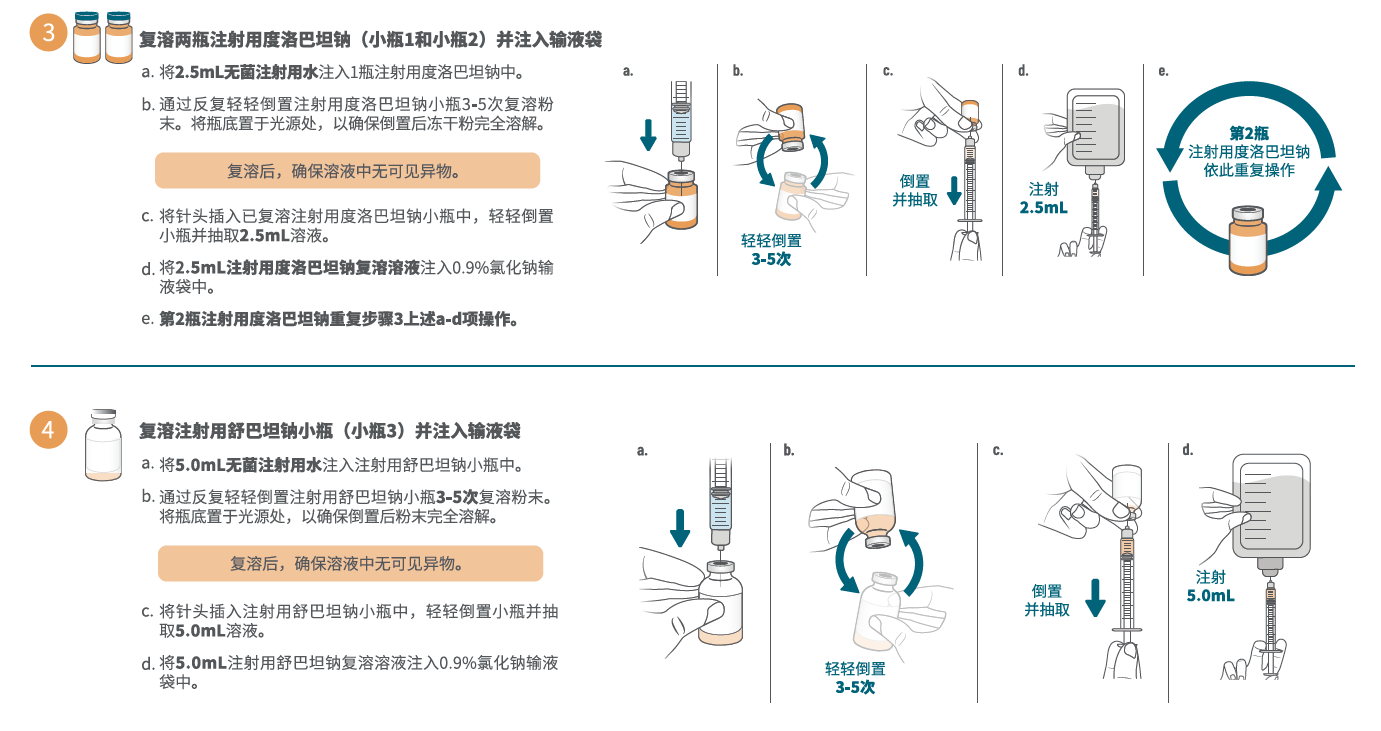
- 加 5.0 mL 无菌注射用水至舒巴坦1.0 g单剂量小瓶,轻轻振摇至溶解。每个溶解小瓶中含 1.0 g 舒巴坦/5.0 mL 澄清、无色至淡黄色溶液。复溶后溶液不能直接注射,必须在静脉输注前稀释。稀释必须在复溶后 1 小时内进行。
- 加 2.5 mL 无菌注射用水至每个度洛巴坦 0.5g 单剂量小瓶,轻轻振摇至溶解。每个溶解小瓶中含 0.5 g 度洛巴坦/2.5 mL 澄清、淡黄色至橙色溶液。复溶后溶液不能直接注射,必须在静脉输注前稀释。稀释必须在复溶后 1 小时内进行。
- 为制备本品所需剂量,分别抽取5.0 mL舒巴坦复溶后溶液和5.0 mL(每小瓶 2.5 mL,共2瓶)度洛巴坦复溶后溶液,向100 mL 0.9% 氯化钠注射液输液袋中注入所抽取体积的舒巴坦和度洛巴坦。本品未用完药液应丢弃。
在溶液和容器允许的情况下,给药前应目视检查注射用药品的颗粒物和变色情况。制备好的本品溶液应为澄清、淡黄色至橙色,不含颗粒物。如果本品溶液浑浊或含有颗粒物,请勿给药。
相容性
本品与 0.9%氯化钠注射液相容。
尚未确定本品与含其他药物或稀释剂的溶液给药相容性。本品不得与其他药物混合或物理添加至含其他药物的溶液中。
以下严重不良反应的详细讨论见【注意事项】:
- 超敏反应(见【注意事项】)
- 艰难梭菌相关性腹泻(CDAD)(见【注意事项】)
临床试验经验
已在 380 例成人受试者中评价了度洛巴坦联合或不联合舒巴坦的安全性,包括 6 项 I 期试验、1 项在复杂性尿路感染(cUTI)患者(包括急性肾盂肾炎)中的 II 期试验和 1 项在鲍曼-醋酸钙不动杆菌复合体所致感染(包括医院获得性细菌性肺炎(HABP)、呼吸机相关性细菌性肺炎(VABP)和通气性肺炎(VP))成人患者中的 III 期试验(也被称为试验 1) (见【临床试验】)。
在 III 期试验的随机、活性对照部分,91 例患者接受本品(1.0 g 舒巴坦和 1.0 g 度洛巴坦,或根据肾功能调整剂量)静脉给药 3 小时,每 6 小时一次,86 例患者在多粘菌素 E 2.5-5 mg/kg 初始负荷剂量给药后接受多粘菌素 E 2.5 mg/kg(或根据肾功能调整剂量)静脉给药 30 分钟,每 12 小时一次。两个治疗组每 6 小时还接受一次 1.0g 亚胺培南/1.0g 西司他丁(或根据肾功能调整剂量)静脉给药,作为鲍曼-醋酸钙不动杆菌复合体以外的潜在 HABP/VABP 病原体的背景治疗。本品治疗的平均持续时间为 9 天,多粘菌素 E 为 8 天。
严重不良反应和终止治疗
本品治疗组的 36 例患者(40%)和多粘菌素 E 治疗组的 42 例患者(49%)发生了严重不良反应。11%(10/91)接受本品治疗的患者和 16%(14/86)接受多粘菌素 E 治疗的患者因出现不良反应而终止治疗。1 例接受本品治疗的患者出现过敏性休克,导致终止治疗。
常见不良反应
本品治疗组 88%(80/91)的患者和多粘菌素 E 治疗组 94%(81/86)的患者报告了不良反应。接受本品治疗患者中最常报告的不良反应(>10%)为肝功能检查异常、腹泻、贫血和低钾血症。表 2 列出了试验 1 中发生率>5%的不良反应。
表 2. 试验 1 中发生率>5%的不良反应
| 不良反应 | XACDURO (N=91) n (%) | 多粘菌素E (N=86) n (%) |
|---|---|---|
| 任何不良反应 | 80 (88) | 81 (94) |
| 肝功能检查异常1 | 17 (19) | 18 (21) |
| 腹泻 | 15 (17) | 9 (11) |
| 贫血 | 12 (13) | 12 (14) |
| 低钾血症 | 11 (12) | 9 (11) |
| 心律失常 | 8 (9) | 8 (9) |
| 急性肾损伤2 | 5 (6) | 31 (36) |
| 血小板减少症 | 5 (6) | 3 (4) |
| 便秘 | 5 (6) | 5 (6) |
1 肝功能检查异常包括以下不良反应:肝功能检查异常、肝功能异常、转氨酶升高、丙氨酸氨基转移酶升高和天门冬氨酸氨基转移酶升高;
2 急性肾损伤包括以下不良反应:肾功能损害、血肌酐升高、中毒性肾病、肾衰和急性肾损伤。
已知对本品成份(舒巴坦和度洛巴坦)或其他 β-内酰胺类抗菌药物有严重超敏反应史的患者应禁用本品(见【注意事项】)。
超敏反应
在接受 β-内酰胺类抗菌药物治疗的患者中曾报告严重和偶发致命性超敏反应(速发严重过敏反应)和严重皮肤反应。这些反应更容易发生在有 β-内酰胺类超敏反应史和/或对多种变应原有过敏史的个体。在本品临床试验中,接受本品治疗的患者有超敏反应发生(见【不良反应】)。在开始本品治疗前,应仔细询问既往对碳青霉烯类、青霉素类、头孢菌素类、其他 β-内酰胺类和其他过敏原的超敏反应史。如果在使用本品时发生过敏反应,应停用本品。
艰难梭菌相关性腹泻(CDAD)
几乎所有抗菌药物(包括本品)的应用都有艰难梭菌相关性腹泻(CDAD)的报告,其严重程度可表现为轻度腹泻至致死性结肠炎。抗菌药物治疗可引起结肠正常菌群的改变,导致艰难梭菌的过度生长。
艰难梭菌产生的毒素 A 和毒素 B 促使 CDAD 的发生和进展。艰难梭菌的高产毒株可导致发病率和死亡率增加,因为抗菌药物治疗可能对这些感染无效,有可能需要结肠切除。对于所有使用抗菌药物后出现腹泻的患者,必须考虑到 CDAD 的可能。由于曾经有给予抗菌药物治疗之后超过 2 个月发生 CDAD 的报道,因此需仔细询问病史。
如果怀疑或确诊 CDAD,应评估继续使用本品治疗的风险/获益。应根据临床指征开始适当的液体和电解质管理、蛋白质补充、艰难梭菌的抗菌治疗以及外科评估。
耐药菌的产生
在未确诊或未高度怀疑细菌感染或作为预防性用药的情况下,本品不太可能对患者产生获益,且有增加耐药菌产生的风险。
孕妇
风险总结
本品
尚无在妊娠期间使用本品的数据用于评估发生重大出生缺陷、流产或其他母体或胎儿不良结局的药物相关风险。
舒巴坦
现有已发表的关于几十年来妊娠期间联合使用舒巴坦与氨苄西林的病例数据,尚未发现重大出生缺陷、流产或其他母体或胎儿不良结局的药物相关风险。已发表的文献报告了舒巴坦可穿过人体胎盘。已在小鼠、大鼠和家兔中以高达人体剂量 10 倍的剂量进行了生殖研究,未显示舒巴坦对胎仔造成伤害的证据(见【药理毒理】)。
度洛巴坦
尚无在妊娠期间使用度洛巴坦的数据用于评估发生重大出生缺陷、流产或其他母体或胎儿不良结局的药物相关风险。
妊娠小鼠和大鼠在器官形成过程中给予度洛巴坦未显示药物诱导的胎仔畸形,但在 2 倍和 4 倍人体最大推荐剂量(MRHD)时观察到小鼠的骨骼变异发生率增加(见【药理毒理】)。
哺乳期妇女
风险总结
尚无关于人体或动物乳汁中存在度洛巴坦的数据。人体乳汁中存在低浓度的舒巴坦。已发表的数据报告,假设婴儿平均乳汁摄入量为 200 mL/kg/天,预计摄入的母乳中舒巴坦最大日剂量为 560 mcg/kg/天(成人体重的 1%至 2%的校正剂量)。没有关于本品、舒巴坦或度洛巴坦对母乳喂养婴儿或乳汁产量影响的数据。
应同时考虑哺乳对婴儿发育和健康的益处、母亲对本品的临床需求以及本品或母亲基础状况对乳母喂养婴儿的任何潜在不良影响。
尚未确定本品在 18 岁以下儿童患者中使用的安全性和疗效。
在试验 1 接受本品治疗的 91 例患者中,49 例(54%)为 65 岁及以上患者,包括 17例(19%)76 岁及以上患者。临床研究没有纳入足够数量 65 岁及以上患者,以确定他们的反应是否与年轻患者不同。
已知本品主要经肾脏排泄,老年患者更容易出现肾功能减退。应基于肾功能调整老年患者的给药方案(见【用法用量】、【临床药理】)。
有机阴离子转运蛋白 1(OAT1)抑制剂
与OAT1 抑制剂合并给药可能会增加舒巴坦的血药浓度。不建议本品与 OAT1 抑制剂(如丙磺舒)合并给药(见【临床药理】)。
尚无与本品滥用和药物依赖相关研究信息。
尚无与本品过量使用相关的临床体征和症状信息。当脑脊液中 β-内酰胺水平升高时,可能发生神经系统不良反应(包括惊厥)。通过血液透析可清除舒巴坦和度洛巴坦(见【临床药理】)。尚无关于使用血液透析治疗用药过量的临床信息。
药效学
对于舒巴坦,已证明未结合血浆舒巴坦浓度高于对鲍曼不动杆菌最小抑菌浓度(MIC)的时间占给药间期百分比(%fT>MIC)是动物和体外感染模型中疗效的最佳预测指标。对于度洛巴坦,24 小时未结合血浆度洛巴坦 AUC 与舒巴坦-度洛巴坦 MIC 的比值(fAUC0–24/MIC)可最佳预测体内和体外感染模型中的活性。
心脏电生理学
在最大推荐单次剂量的 4 倍剂量下,度洛巴坦未引起任何临床相关程度的 QTc 间期延长。
药代动力学
舒巴坦和度洛巴坦单次和多次给药后的药代动力学相似。舒巴坦的 Cmax 和 AUC 随剂量成比例增加(0.5 倍推荐单次给药剂量至 1.0 g 单次给药剂量)。度洛巴坦在研究的剂量范围内(0.25 倍推荐单次给药剂量至 2 倍推荐单次给药剂量,每 6 小时输注 1 次,每次持续 3 小时)显示出与剂量成比例的药代动力学。
本品各组分药代动力学特征以及舒巴坦和度洛巴坦的药代动力学参数总结见表 3。
表 3:本品各组分的药代动力学特征(平均值±SD)
| 药代动力学参数 | 舒巴坦 | 度洛巴坦 |
|---|---|---|
| Cmax (µg/mL)* | 32.4 ±24.7 | 29.2 ±13.2 |
| AUC0-24 (h·µg/mL )* | 515 ±458 | 471 ±240 |
| 分布 | ||
| 与人血浆蛋白结合百分比 | 38% | 10% |
| AUC0-6 ELF/血浆比率 | 0.5 | 0.37 |
| Vss (L)* | 25.4 ±11.3 | 30.3 ±12.9 |
| 代谢 | 代谢极少 | |
| 消除 | ||
| CL (L/h)* | 11.5 ±5.64 | 9.96 ±3.11 |
| T1/2(h)* | 2.15 ±1.16 | 2.52 ±0.77 |
| 排泄 | ||
| 主要消除途径 | 肾脏 | |
| 原形经尿液排泄的百分比 | 75%-85% | 78% |
* 显示数值为在给药剂量为 1.0 g 舒巴坦和 1.0 g 度洛巴坦每 6 小时一次的肾功能正常(定义为肌酐清除率≥90 mL/min 且<130 mL/min)患者中的稳态(第 3 天)药代动力学参数。
AUC0-6 = 从给药时至给药后 6 小时的血浆浓度时间曲线下面积;AUC0-24 = 从给药时至 24 小时的血浆浓度时间曲线下面积;CL = 清除率;Cmax = 峰浓度;ELF = 上皮细胞衬液;SD = 标准差,T1/2= 终末半衰期;Vss = 稳态分布容积
特殊人群
基于年龄(18-91 岁)、性别、体重(35-150 kg)和人种(白人、黑人、亚洲人、美国印第安人/阿拉斯加原住民、其他),未观察到舒巴坦或度洛巴坦具有临床意义的药代动力学差异。
肾功能损害患者
在一项评价肾功能损害对舒巴坦和度洛巴坦药代动力学影响的单次给药试验中,与 CLcr≥90 mL/min 的健康受试者相比,所有肾功能损害水平下舒巴坦和度洛巴坦的剂量标准化的全身暴露量均较高(表 4)。在接受血液透析的终末期肾脏疾病(ESRD)受试者中,舒巴坦和度洛巴坦通过血液透析清除的剂量分数分别为 0.41 和 0.33。
表 4:与 CLcr≥90mL/min 的受试者相比,肾功能损害受试者剂量标准化的 AUC 增加倍数
| 估计 eGFR(mL/min/1.73 m2) | 舒巴坦 | 度洛巴坦 |
|---|---|---|
| ≥60 且<90 | 1.4 | 1.4 |
| ≥30 且<60 | 2.0 | 1.9 |
| <30 | 4.3 | 3.7 |
为维持与肾功能正常患者相似的全身暴露量,建议肾功能损害患者调整剂量。接受血液透析的患者应在血液透析完成后接受本品治疗(见【用法用量】)。
CLcr≥130 mL/min 的患者
在 CLcr≥130 mL/min 的患者中观察到舒巴坦和度洛巴坦清除率升高。每 4 小时输注一次本品(1.0g 舒巴坦和 1.0g 度洛巴坦),每次持续 3 小时,舒巴坦和度洛巴坦暴露量与 CLcr 为 90-129 mL/min 的患者暴露量相当(见【用法用量】)。
肝功能损害患者
舒巴坦和度洛巴坦主要经肾脏清除;因此,肝功能损害不太可能对本品暴露量产生任何影响(见【用法用量】)。
药物相互作用研究
临床研究
一项在健康受试者中开展的临床研究,未观察到度洛巴坦、舒巴坦、亚胺培南和西司他丁之间的药物-药物相互作用。
未对药物相互作用潜力进行进一步临床评价的体外研究:在治疗性血浆浓度下,度洛巴坦不抑制 CYP1A2、CYP2A6、CYP2B6、CYP2C8、CYP2C9、CYP2C19、CYP2D6、CYP2E1 或CYP3A4。度洛巴坦无体外诱导 CYP1A2、CYP2B6 或CYP3A4 的潜力。
未开展 CYP450 和舒巴坦的体外研究。
膜转运系统 :
体外数据显示,在治疗性血浆浓度下,舒巴坦不抑制 P-gp、BCRP、OATP1B1、OATP1B3、OCT1、OCT2、BSEP、OAT1、OAT3、MATE1 或 MATE2-K。体外数据还表明,在治疗性血浆浓度下,度洛巴坦不抑制 P-gp、BCRP、OATP1B1、OAT1、OAT3或 OCT2。舒巴坦和度洛巴坦均为 OAT1 的底物。但是,预计主动分泌作用仅为舒巴坦总清除率的重要部分,因此,抑制 OAT1 可能会增加舒巴坦的血浆浓度。尚未开展本品和 OAT1 抑制剂的临床研究。(见【药物相互作用】)。
鲍曼-醋酸钙不动杆菌复合体所致医院获得性和呼吸机相关性细菌性肺炎的治疗
在一项多中心、活性对照、研究者非盲、独立评估者设盲、非劣效性 III 期试验(试验 1,NCT03894046)中,共 177 例证实为鲍曼-醋酸钙不动杆菌复合体感染的住院成人患者接受了随机化和治疗。患者接受本品(1.0 g 舒巴坦和 1.0 g 度洛巴坦,或根据肾功能调整的剂量)静脉给药 3 小时,每 6 小时一次(n=91)或在多粘菌素 E 2.5-5 mg/kg 初始负荷剂量给药后,接受多粘菌素 E 2.5 mg/kg(或根据肾功能调整的剂量)静脉给药 30分钟,每 12 小时一次(n=86)。两个治疗组均接受每 6 小时一次的 1.0 g 亚胺培南/1.0 g西司他丁(或根据肾功能调整的剂量)静脉给药,作为鲍曼-醋酸钙不动杆菌复合体以外的潜在 HABP/VABP 病原体背景治疗。患者接受最多 14 天的治疗。
本研究的主要疗效终点是接受任何数量研究药物且证实基线感染碳青霉烯类耐药的鲍曼-醋酸钙不动杆菌复合体(CRABC 微生物学改良意向治疗(m-MITT)人群)的患者的 28 天全因死亡率。在 CRABC m-MITT 人群的 128 例患者中,评估了 125 例在第 28 天生存状态评估前未撤回知情同意的患者:本品组 63 例患者和多粘菌素 E 组 62例患者。
在分析的 125 例患者中,治疗组之间的人口统计学和基线特征具有可比性:74%为男性, 50%为白人,44%为亚洲人,平均年龄为 63(±17)岁。大多数患者的基线感染类型为肺炎(53% VABP 和 43% HABP),2%为菌血症。基线时的平均急性生理学和慢性健康评价 II(APACHE II)评分为 17,26%的患者的 APACHE II 评分≥20。随机化时, 64%的患者在 ICU 中≥5 天;26 % 的患者在 ICU 中>14 天,75 % 的患者进行过机械通气。约 39%的患者在基线时的 CLcr<90 mL/min。本品治疗组的平均治疗持续时间为 9 天,多粘菌素 E 组为 8 天。
表 5 显示了主要终点(CRABC m-MITT 人群 28 天全因死亡率)的结果。在 28 天全因死亡率方面,本品非劣效于多粘菌素E。
表5:第28 天时的全因死亡率(CRABC m-MITT 人群)
| 本品 n/N* (%) | 多黏菌素 E n/N* (%) | 治疗差异 (95% CI) † | |
|---|---|---|---|
| 28 天全因死亡率 | 12/63 (19%) | 20/62 (32.3%) | -13.2% (-30.0, 3.5) |
CI=置信区间;
*使用指定人群中的患者人数 N 作为分母计算百分比。
†使用连续性校正的 z 统计量计算 95% CI(双侧)。
研究还评价了临床治愈率。临床治愈定义为:基线时存在的体征和症状痊愈或显著改善,无新症状,因此无需其他抗革兰阴性抗菌治疗。治愈检验(TOC)访视时,即治疗结束后 7 天(±2 天),CRABC m-MITT 人群中本品试验组的临床治愈率为 39/63(61.9%),多粘菌素E组为 25/62(40.3%)。
药理作用
注射用舒巴坦钠/注射用度洛巴坦钠组合包装产品为抗菌药物,含有舒巴坦和度洛巴坦。
作用机制
舒巴坦是一种 β-内酰胺类抗菌药物和 Ambler A 类丝氨酸 β-内酰胺酶抑制剂,因可抑制鲍曼-醋酸钙不动杆菌复合体(ABC)青霉素结合蛋白 PBP1 和PBP3(细菌细胞壁合成所需的必需酶)而具有抗菌活性。
度洛巴坦是一种二氮杂二环辛烷、非 β-内酰胺类的 β-内酰胺酶抑制剂,可保护舒巴坦不被 β-内酰胺酶降解。度洛巴坦单独给药对 ABC 分离株没有抗菌活性。
已证实本组合包装产品对表达丝氨酸 β-内酰胺酶(包括 Ambler A 类(CTX-M-、 TEM-、PER-和 SHV 型超广谱 β-内酰胺酶[ESBL]、KPC 碳青霉烯酶)、C 类(ADC 型))的 ABC 分离株具有体外活性,对 D 类(OXA 型)酶具有广谱活性。
耐药性
ABC 分离株对 β-内酰胺类抗菌药物产生耐药性的机制可能包括产生 β-内酰胺酶、 PBP 修饰或靶点改变、外排泵上调或外膜孔蛋白丢失。
本组合包装产品对产生 Ambler B 类金属 β-内酰胺酶或舒巴坦活性靶点(即 PBP)经修饰的 ABC 分离株没有活性。分离株还可能同时产生多种 β-内酰胺酶、表达不同水平的 β-内酰胺酶、具有氨基酸 PBP 序列变异或其他可能导致耐药性的耐药机制。
与其他抗菌药物的相互作用
体外研究显示,本组合包装产品与其他抗菌药物(包括头孢他啶-阿维巴坦、亚胺培南、美罗培南、阿米卡星、多粘菌素 E、头孢吡肟、环丙沙星、米诺环素、利福平、利福昔明、达托霉素、达巴万星、奥利万星、特地唑胺、非达霉素、万古霉素、利奈唑胺、甲硝唑和氟康唑)之间无拮抗作用。
动物感染模型
在动物感染模型(包括中性粒细胞减少小鼠的大腿和肺部感染)中,度洛巴坦恢复了舒巴坦对本组合包装产品敏感性鲍曼不动杆菌菌株的活性。
抗菌活性
在体外和临床感染中,已证明本组合包装产品对以下微生物的大多数分离株具有活性。鲍曼-醋酸钙不动杆菌复合体
药敏试验
在可能的情况下,临床微生物实验室应定期向医生报告并提供其所在医院使用的抗菌药物的相关体外药敏性试验结果,该结果可反映医院及社区获得性病原菌的药敏特点。这些报告可帮助医生在治疗时选择抗菌药物。
稀释法
通常采用定量方法测定抗菌药物的最小抑菌浓度(MIC)值。MIC 用于评估细菌对抗菌化合物的敏感性。应使用标准化的实验方法(微量肉汤稀释法)测定本品 MIC。舒巴坦联合固定浓度 4μg/mL 度洛巴坦的 MIC 值应通过系列稀释确定。应根据表 6 中的标准对 MIC 进行判读。
扩散法
通过测量抑菌圈直径的定量法也可用于重复估计细菌对抗菌药物的敏感性。应使用标准化的实验方法测定抑菌圈直径。使用含有 10μg 舒巴坦和 10μg 度洛巴坦的纸片检测微生物对本品的敏感性。纸片扩散法判读标准见表 6。
表6 舒巴坦-度洛巴坦药敏试验结果判读标准
| MIC (mcg/mL) | 纸片扩散法抑菌圈直径 (mm) | |||||
|---|---|---|---|---|---|---|
| 病原体 | S | I | R | S | I | R |
| 鲍曼-醋酸钙 不动杆菌复合体 | ≤ 4/4 | 8/4 | ≥ 16/4 | ≥ 17 | 14 - 16 | ≤ 13 |
S = 敏感; I = 中介; R = 耐药
报告结果为“敏感(S)”时,表示如果抗菌药物在感染部位达到通常浓度时,很可能会抑制病原体的生长。报告结果为 “中介( I )”时,表示细菌引起的感染仅在应用高剂量抗菌药物时有效,或者细菌处于体内抗菌药物浓缩的部位或体液中时才被抑制。报告结果为 “耐药(R)”时,表示即使抗菌药物在感染部位达到通常浓度,也可能无法抑制病原体生长,应选择其他疗法。
质量控制
标准化药敏试验程序需要采用实验室质控措施,以监测并确保本试验所用材料和试剂的准确度和精密性,以及试验执行人员的操作方法正确。标准的本品粉末采用微量肉汤稀释法应能得出表7 列出的MIC值范围。使用10μg舒巴坦和10μg度洛巴坦纸片扩散法应该能达到下表7 所示的标准。
表 7:舒巴坦-度洛巴坦药敏试验可接受的质量控制范围
| 参比菌株 | MIC 范围(μg/mL)a | 纸片扩散法 (抑菌圈直径范围,mm) |
|---|---|---|
| 鲍曼不动杆菌 NCTC 13304 | 0.5/4- 2/4 | 24 - 30 |
a. 舒巴坦联合固定浓度 4 μg/mL 度洛巴坦的 MIC。
毒理研究
遗传毒性
度洛巴坦细菌回复突变试验、体内和体外微核试验以及大鼠体内 Pig-A 试验结果均为阴性。
未对舒巴坦或舒巴坦-度洛巴坦组合开展遗传毒性研究。
生殖毒性
舒巴坦:
在小鼠、大鼠和兔中以高达人体剂量 10 倍的剂量开展的生殖研究中,未见舒巴坦钠/氨苄西林钠对胎仔的损伤。
度洛巴坦:
雄性和雌性大鼠静脉给予度洛巴坦剂量高达 1000 mg/kg/天(以 AUC 计,约为人最大推荐剂量(MRHD)的 4 倍),雌性小鼠皮下给予度洛巴坦剂量高达 1600 mg/kg/天(以 AUC 计,相当于 MRHD 的 4 倍),均未观察到对生育力、生殖能力、胎仔存活、生长或产后发育的不良影响。
妊娠小鼠自妊娠(GD)第 6 天至第 15 天,皮下注射给予度洛巴坦 400、800 或1600mg/kg/天(每天给药 4 次),800 和/或 1600 mg/kg 剂量(以 AUC 计,相当于 MRHD 的 2 和 4 倍)下可见度洛巴坦相关的后肢骨骼变异(跟骨未骨化)、胸骨不对称骨化和多余肋骨发生率增加。各剂量组均未见对平均体重、生殖能力或剖宫产参数的不良影响。
妊娠 SD 大鼠自 GD6 至哺乳期第 20 天,静脉输注度洛巴坦(2 小时/天)300 或 1000 mg/kg/天(以 AUC 计,约为 MRHD 的 4 倍),耐受性良好,两组中均未见母体不良反应。各剂量组均未见对胚胎-胎仔及产前产后发育的不良影响。
致癌性
尚未开展本组合包装产品的致癌性研究。
密闭,2~8℃保存。
请将本品放在儿童不能接触的地方。
注射用舒巴坦钠:中硼硅玻璃模制注射剂瓶(无色)、注射液和注射用无菌粉末用溴化丁基橡胶塞。
注射用度洛巴坦钠:中硼硅玻璃管制注射剂瓶(棕色)、注射制剂用氯化丁基橡胶塞。
每盒含注射用舒巴坦钠(1.0g,按 C8H11NO5S 计)1 瓶与注射用度洛巴坦钠(0.5g,按 C8H11N3O6S 计)2 瓶。
24个月
JX20240032
国药准字HJ20240024
名称:Entasis Therapeutics, Inc.
注册地址:930 Winter St - Suite 1500, Waltham, MA 02451 United States of America
组合包装中注射用舒巴坦钠
企业名称:Mitim S.r.l.
生产地址:Via Gian Battista Cacciamali 34, Brescia, Brescia 25125 ITALY
组合包装中注射用度洛巴坦钠
企业名称:Lyophilization Services of New England, Inc. (LSNE)
生产地址:25 Commerce Drive, Bedford, NH 03110 United States of America
企业名称: 再鼎医药(上海)有限公司
注册地址:中国(上海)自由贸易试验区哈雷路899号1幢B座1-3层、5-8层
产品咨询电话:400-820-1022
2024年5月15日
2024年12月19日;2025年06月22日



