请仔细阅读说明书并在医师指导下使用
血友病 A 患者出血的控制和预防
本品适用于血友病 A(先天性凝血因子Ⅷ缺乏)患者出血的控制和预防。
本品不含血管假性血友病因子,因此不适用于治疗血管性血友病(von Willebrand disease,vWD)。
血友病 A 患者的手术预防
本品适用于血友病 A 患者的手术出血预防。
250 IU/瓶、500 IU/瓶、1000 IU/瓶、2000 IU/瓶。
用药剂量
本品应在有血友病 A 患者治疗经验的医师指导下使用。
本品的剂量和治疗持续时间取决于患者因子Ⅷ缺乏的严重程度、出血的部位与范围以及患者的临床状况。应根据患者的临床反应调整给药剂量。在大手术或危及生命的出血事件中,对本品的治疗进行监控尤为重要。
本品所标示的一个国际单位 (IU)的因子Ⅷ活性大约相当于 1 ml 正常人血浆中因子Ⅷ的含量。所需因子Ⅷ剂量的计算基于实践经验,即每 kg 体重的 1 IU 因子Ⅷ平均可使血浆因子Ⅷ的活性升高约 2 IU/dL。可用下面的公式计算所需剂量:
需要剂量(IU)= 体重(kg) × 因子Ⅷ期望升高值(IU/dL 或% )× 0.5 [(IU/kg)/(IU/dL)]
对下列出血的控制和手术预防,根据以下的出血分类,因子Ⅷ活性不应低于对应时期内规定的血浆活性水平(按正常的% 或IU/dL)。
表1. 控制出血和手术预防的给药剂量指导
| 出血类型 | 所需因子Ⅷ水平(%) | 给药频率(小时)/ 治疗持续时间(天) |
|---|---|---|
| 少量出血 | ||
| 早期关节积血、浅表肌肉或软组织和口腔出血 | 20~40 | 根据需要,每 12~24 小时重复给药,直至缓解。根据出血的严重程度,至少治疗 1 天。 |
| 中度出血和小手术 | ||
| 中度肌肉内出血;轻度头部外伤。小手术,包括拔牙。口腔出血。 | 30~60 | 每 12~24 小时重复注射,治疗 3~4 天,或直到止血和伤口愈合。在拔牙时,1 小时内接受单次注射加口服抗纤溶药物可能足够。 |
| 大出血和大手术 | ||
| 胃肠道出血;颅内、腹腔或胸腔内出血;骨折。大手术。 | 60~100 | 每 8~24 小时重复注射,直至危险消除,或止血和手术伤口愈合。 |
建议用测定因子Ⅷ活性的方法监测本品的因子活性,尤其对于外科手术的干预。临床数据支持使用一期凝固法监测因子Ⅷ的活性。
根据每个血友病 A 患者当前的治疗方案,应建议在旅行时携带充足的因子Ⅷ产品,以满足治疗需要。应建议患者在旅行之前咨询医生。
使用说明
使用本品包装中所提供的预装注射器中的稀释剂(0.9 %氯化钠溶液)复溶冻干粉后,进行静脉注射给药。
配制:
- 在进行下列操作前须洗净双手。
- 复溶时采用无菌操作技术(即洁净、无菌)。
打开无菌包装后,尽快使用复溶和给药所需的所有物品,尽量减少在空气中不必要的暴露。
注意:如果注射时需要使用1瓶以上的本品,按下述方法对每瓶药物进行复溶。应移除稀释液注射器,将接合器留在药瓶上,可另用1支一次性无菌注射器,如:大容量的路厄旋扣注射器(luer lock syringe),抽取每瓶内的复溶物。直到抽取下一瓶药物时,才可把待抽取药物的稀释液注射器移除,并将大容量路厄旋扣注射器从上一瓶药物中取出插入待抽取药物瓶中。
复溶:
- 需将本品瓶内的冻干粉和预装稀释液的注射器放置至室温。
移除药瓶上的塑料盖,露出胶塞的中心部位。
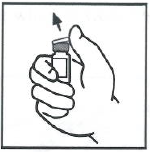
- 用酒精棉或其它消毒溶液擦拭药瓶顶部,自然晾干。消毒后,切勿用手触摸胶塞,并防止胶塞接触任何物体。
- 揭开接合器的透明塑料包装保护套。不要将接合器从包装内取出。
将药瓶放在一个平整的台面上。握住包装内的接合器,放在药瓶上方。用力下压包装,直到接合器的针头刺入瓶塞内。
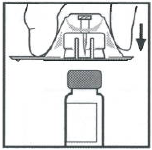
如图所示,抓住稀释液注射推杆底部,并通过严实推压及旋转将注射器推杆的螺纹端装入注射器柱塞中。在操作过程中避免接触注射器的推杆体部。
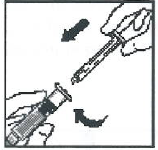
折断注射器护套的穿孔处,从注射器上折下防启塑料尖嘴护套。不要触摸护套的内侧或注射器尖部。如果复溶后的本品不立即使用,由于稀释液注射器可能需要用护套重新盖住,因此需将护套倒置放在一个干净、最不易被环境污染的台面上。
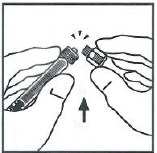
提起接合器的塑料包装并丢弃包装。
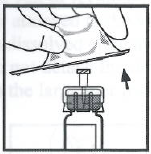
将药瓶放在平整的台面上。将注射器尖部插入接合器开口处,顺时针用力旋转推动注射器直到锁定,以便将稀释液注射器连接在接合器上。
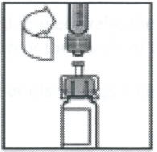
缓慢下压注射器推杆,将所有稀释液注入药瓶内。
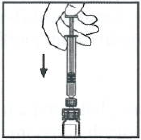
保持注射器与接合器相连接,轻轻摇晃药瓶内的液体,直到粉末溶解。
注意:注射前应肉眼检查最终溶液有无颗粒物质;此溶液应为无色澄清至淡乳白色,否则应丢弃另使用新的本品。
倒置药瓶,缓慢将溶液吸入注射器内。
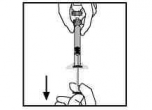
缓缓地逆时针旋转拔出注射器,将注射器与接合器分离。丢弃药瓶与接合器。
注意:如果不立即使用,应小心盖上注射器护套。不要触摸注射器尖部或注射器护套的内侧。
给药前,将复溶溶液在室温下储存,并在 3 小时内使用。
本品复溶后含有聚山梨酯 80,已知其可增加邻苯二甲酸二-(2-乙基己基)盐(DEHP)从聚氯乙烯(PVC)中的提取。这一点在本品的配制和给药(包括在 PVC 容器中的保存时间)过程中应予以注意。应严格按照【用法用量】中的建议使用本品。
给药:
使用本品包装中提供的预装注射器内稀释液(0.9 %氯化钠溶液,4 ml)复溶本品后,静脉注射给药。给药前应检查本品中有无颗粒物及是否变色。
注射本品时,应使用本品包装中所提供的静脉输液针和预装稀释液注射器,或一次性无菌注射器(如大容量的路厄旋扣注射器)。此外,应使用所附接合器从药瓶中抽吸溶液。
- 将注射器连接到包装中所附的静脉输液针的螺旋接口端。
扎好止血带,并用酒精棉擦拭皮肤注射部位。
进行静脉穿刺。将静脉输液针的针头刺入静脉内,取下止血带。本品的复溶溶液应该在几分钟内完成静脉注射。根据患者的舒适程度调节注射速度。
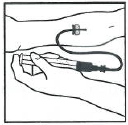
复溶后的本品不能与其它药品共用同一导管或容器。
本品注射完毕后,取下并丢弃静脉输液针。将所有未使用的溶液、空瓶及使用过的针头和注射器弃入合适的容器内,以防处理不当伤及他人。
安全信息汇总
使用本品的患者偶见超敏反应或过敏反应(可能包括血管性水肿、注射部位烧灼感和刺痛感、寒战、潮红、全身性荨麻疹、头痛、荨麻疹、低血压、嗜睡、恶心、躁动、心动过速、胸闷、刺痛感、呕吐、哮鸣),在某些情况下可能发展成为包括休克在内的速发严重过敏反应(参见【警告和注意事项】)。
如果发生疑似与本品有关的不良反应,应根据患者的表现降低静脉注射速度或终止注射本品。
本品可能存在微量仓鼠蛋白。在极罕见情况下观察到仓鼠蛋白抗体形成,但不存在临床后遗症。在一项研究中,113 位既往接受过治疗的患者(PTP, previously treated patients)中,有 20 位(18%)曾出现抗中国仓鼠卵巢细胞(CHO)抗体滴度升高,并且无任何明显的临床效果。
使用包括本品在内的因子Ⅷ治疗的血友病 A 患者可能会出现中和抗体(抑制物)。应监测抑制物形成情况。若形成抑制物,患者可能会出现临床疗效不佳。在此情况下,建议与血友病专科中心取得联系。
不良反应列表
按照MedDRA系统器官分类(SOC和首选术语级别),不良反应发生率按以下评估:很常见(≥ 1/10);常见(≥ 1/100至< 1/10)和少见(≥ 1/1,000至< 1/100)。下表列出本品临床试验中报告的不良反应。包括765名受试者的汇集临床试验治疗中出现的全因性不良事件。以下不良反应发生率按严重程度递减顺序列出。
表2. 本品的不良反应汇总
| 系统器官分类 | 很常见 ≥ 1/10 | 常见 ≥ 1/100 至 < 1/10 | 少见 ≥ 1/1,000 至 < 1/100 |
|---|---|---|---|
| 血液及淋巴系统疾病 | 因子Ⅷ抑制(PUP)* | 因子Ⅷ抑制(PTP)* | |
| 免疫系统疾病 | 速发严重过敏反应** | ||
| 代谢及营养类疾病 | 食欲减退 | ||
| 各类神经系统疾病 | 头痛 | 头晕 | 周围神经病、嗜睡、味觉倒错 |
| 心脏器官疾病 | 心绞痛、心动过速、心悸 | ||
| 血管与淋巴管类疾病 | 出血、血肿 | 低血压、血栓性静脉炎、潮红 | |
| 呼吸系统、胸及纵膈疾病 | 咳嗽 | 呼吸困难 | |
| 胃肠系统疾病 | 腹泻、呕吐、腹痛、恶心 | ||
| 皮肤及皮下组织类疾病 | 荨麻疹、皮疹、瘙痒症 | 多汗 | |
| 各种肌肉骨骼及结缔组织疾病 | 关节痛 | 肌痛 | |
| 全身性疾病及给药部位各种反应 | 发热 | 寒战、插管部位相关反应 | 乏力、注射部位反应、注射部位痛、注射部位炎症 |
| 各类检查 | 抗体检查阳性、抗因子Ⅷ抗体检查阳性 | 天门冬氨酸氨基转移酶升高、丙氨酸氨基转移酶升高、血胆红素升高、血肌酸磷酸激酶升高 |
* 抑制物发生频率采用原产地欧盟《人血浆源性和重组凝血因子 Ⅷ产品核心 SmPC 指南第 3 版》相关内容,这是在重型血友病A患者中研究所有因子Ⅷ产品的结果综合确定的。PTP = 既往接受过治疗的患者,PUP = 既往未接受过治疗的患者
** 本说明书“速发严重过敏反应”均依据 MedDRA 中文编码,下同。
因子Ⅷ抑制
既往接受过治疗的患者
在包含 591 名接受了本品前代产品(1 项临床研究)或本品(6 项临床研究)的 PTP 的汇总数据集中,有 10(1.7%)例确诊的因子Ⅷ抑制物病例(1 例高滴度(≥ 5 BU/mL),9 例低滴度(< 5 BU/mL))。
在针对既往接受过治疗的患者(PTP)(因子Ⅷ:C ≤ 2%)开展的本品临床研究(关键的 3 期临床研究--研究 310)中,因子Ⅷ抑制物的发生率是主要安全性终点。经治的血友病 A 患者用本品进行预防性治疗和按需治疗时, 其中 94 例受试者使用了至少 1 次本品,注射总次数为 6775 次。有 2 例患者出现无临床症状的低滴度一过性抑制物:在暴露天数(ED,范围为 1-92 天)的中位数为 76 天的 94 位患者中,有两例患者出现无临床症状的低滴度一过性抑制物,而在至少暴露 50 天的 89 位患者中,发生率为 2.2%。
在本品的支持研究(研究 310)中,在 110 位患者(PTP)(因子Ⅷ:C ≤ 2%)中观察到 1 例新发抑制物和 2 例复发性抑制物(均为低滴度,中心实验室测定);ED 的中位数为58天(范围为 5-140 天),98 位患者至少暴露于本品50天。在最初的 110 位患者中,有九十八(98)位继续在第二项支持研究中接受治疗,并在随后延长了本品暴露时间,额外 ED 的中位数为 169 天(范围为9-425天)。观察到另外 1 例低滴度新发抑制物。
在针对接受大型手术的血友病 A 的 PTP(因子Ⅷ:C ≤ 2%)的临床研究中,在接受本品治疗的 30 位患者中出现 1 例低滴度抑制物。
在对 PTP(因子Ⅷ:C < 2%)的本品临床研究中,在 113 位患者中观察到 1 例高滴度抑制物。
在儿童(年龄<12 岁,N=37)PTP(因子Ⅷ:C <1%)中进行的本品临床研究中,出现有临床意义的抑制物的患者百分比是主要安全性终点。没有患者达到方案规定的有临床意义的因子Ⅷ抑制标准。观察到 2 例患者(年龄<6 岁)出现暂时性低滴度因子Ⅷ抑制物。两例患者在同一次访视中(ED 10-15)显示回收率下降,抑制物检测为阳性,之后恢复到预期的回收率。两例患者都没有因子Ⅷ抑制物的临床表现,没有因为该事件接受特殊治疗。
在自发性上市后报告中,有 PTP 产生高滴度抑制物的报告。
在暴露天数至少为 50 天的 144 例成人和儿童经治患者中评估了使用本品后因子 Ⅷ 抑制物的生成情况。在临床研究中,对实验室检查 因子Ⅷ 抑制物(部分 Nimegen 改进的 Bethesda 抑制物测定)的结果进行了评估。阳性因子Ⅷ 抑制物的诊断标准为 ≥ 0.6 BU/mL。在所有研究中,3 例(2.1%)受试者出现了因子 Ⅷ 抑制物。
所有研究中,在 40 例不满 16 岁(25 例不满 6 岁以及 15 例 12 至 16 岁)且暴露天数至少为 50 天的经治儿童患者中评估了安全性。在这些患者中,1 例儿童受试者出现了抑制物。
既往未接受过治疗的患者(PUP)
在一项临床试验中(研究 301),在 101 例 PUP 中使用本品前一代产品治疗后有 32 例(32%)出现抑制物:101 例中有 16 例(16%)为高滴度(>5 BU/mL),有 16 例(16%)为低滴度(≤5 BU/mL)。这些患者中出现抑制物前的暴露天数中位值为 12 天(范围:3~49天)。在 16 例高滴度患者中,15 例接受了免疫耐受诱导(ITI),在 16 位低滴度患者中,其中 10 位已开始 ITI。
一项在 PUP(6 岁以下,n=23)中开展的本品临床研究(研究 4434)中,8 名(34.8%)患者出现因子Ⅷ抑制物(4 名患者为高滴度>5 BU/mL,4 名患者为低滴度≤5 BU/mL)。其中 5 名(21.7%)患者符合方案规定的有临床意义的因子Ⅷ抑制物标准,即连续两份血样的抑制物检测结果呈阳性,需要使用其他止血产品和/或因子Ⅷ恢复水平低,以及疗效不佳。
抑制物发生率的贝叶斯(Bayesian)统计分析
在贝叶斯统计分析中,根据研究 310的试验结果,对本品在 PTP 中的支持性试验结果进行了更新。在该试验中,110 例受试者中观察到 1 例新发抑制物,2 例复发性抑制物。此贝叶斯分析显示,该试验人群(真实的)抑制物发生率的 95 %上限的估计值为4.17 %(见表3)。
表3. 抑制物发生率的贝叶斯(Bayesian)后验分布
| 因子Ⅷ 抑制物Nijmegen 结果 (BU/mL) | 抑制物数量 | 接受分析的 受试者数量 | 观察到的抑制物 发生率(%) | β后验分布特点 | |||
|---|---|---|---|---|---|---|---|
| α a | β b | 后验概率c | 抑制物发生率的 95%上限 (%)d | ||||
| ≥ 0.6 | 2 | 89 | 2.25 | 4.5 | 197 | 0.9613 | 4.17 |
注:a 前验α: 2.5+观察到的出现抑制物的受试者数量。
b 前验β :110+接受分析的受试者数量-观察到抑制物的受试者数量。
c 后验概率指的是:真实的抑制物发生率低于可接受上限(4.4%)的概率。后验概率超过0.95即认为可以接受。
d 基于后验分布得出的真实的抑制物发生率的95%上限(在至少95%的概率基础上,计算了最大比率)。低于4.4 %的抑制物发生率是可以接受的。
免疫原性
临床试验期间和上市后收到的疗效不佳的报告主要来源于预防性治疗的患者。产生了抑制物的患者曾报告过疗效不佳和/或因子Ⅷ活性回收率不佳的事件,但未发现抑制物的患者也出现过这些情况。疗效不佳表现为:特定关节出血、新发现的关节出血、其他出血或患者发现的新部位自发性出血。为了确保疗效,必须对每例患者进行个体化的剂量调整并进行监测,尤其是在接受本品治疗的初期。
已在对因子Ⅷ产生抑制物的血友病 A 患者中收集了关于免疫耐受诱导(ITI)的数据。作为对PUP使用本品的关键性试验的一部分,审查了 25 位患者(15 位高滴度,10 位低滴度)的 ITI 数据。在这 25 位患者中,20 位患者的抑制物滴度降低至< 0.6 BU/mL,其中在 15 位最初为高滴度(≥ 5 BU/mL)的患者中,有 11 位滴度下降;10 位最初为低滴度的患者中,9 位滴度下降。因此, 免疫耐受诱导的总体有效性为 80%(20/25),高应答者为 73%,低应答者为 90%。在 6 位产生低滴度抑制物但未接受ITI的患者中,5 位出现类似的滴度降低。目前尚无长期结果。
儿科人群
据报告,一位 11 岁患者出现囊肿,一位 13 岁患者出现意识模糊,这些事件可能与本品治疗有关。
在成人 PTP 以及儿童和青少年 PTP 的研究中评估了本品的安全性(在一项研究中,n=18,患者年龄为 12-16 岁;在一项支持研究中,n=49,患者年龄为 7-16 岁),与成人相比,7-16 岁儿童的不良反应频率有增加的趋势。在包含 PTP(n=18,年龄<6 岁;n=19,年龄 6-<12 岁)和 PUP(n=23,年龄<6 岁)的儿童患者中获得额外的儿童安全性数据,证实其安全性特征与成人患者相似。所有研究中,在 48 例不满 16 岁(28 例不满 6 岁以及 20 例 12 至 16 岁)的经治儿童患者中评估了安全性。共进行了 7,150 次注射,每次注射的剂量中位值为 29 IU/kg(最小值:9 IU/kg,最大值:108 IU/kg)。
所有研究中,成人和儿童 PTP 使用本品时最常见不良反应(≥ 10%)有头痛(26%)、关节痛(25%)、发热(21%)和咳嗽(11%)。5% 或以上受试者报告的其他不良反应有腹泻(8%)、呕吐(7%)、乏力(7%)和恶心(6%)。
上市后经验
本品上市后报告的不良反应:速发严重过敏反应和疗效不佳。
因自发报告上述不良反应的例数不确定,故尚不能确切评估这些不良反应的发生频率。
对本品任何成份过敏者禁用。
本品禁用于已知对仓鼠蛋白有超敏反应史的患者。
1. 一般注意事项
患者对本品的临床反应可能存在个体差异。若使用推荐的剂量未控制出血,应测定血浆中凝血因子Ⅷ水平,并给予足够剂量的本品,以获得满意的临床疗效。若患者血浆因子Ⅷ水平未达到预期,或给予预期剂量后出血未控制,应考虑是否存在抑制物(中和抗体),并做相应检测。
2. 超敏反应
使用本品可能发生超敏反应(包括速发严重过敏反应)。应当将超敏反应和速发严重过敏反应的早期症状告知患者,包括荨麻疹[皮疹伴瘙痒]、全身性荨麻疹、胸部紧压感、哮鸣和低血压。
如果发生过敏性反应,应当立即停用本品,根据反应的种类和严重程度,给予适当的紧急医疗处理,包括休克的治疗。
3. 抑制物
使用凝血因子Ⅷ产品的患者可能出现中和抗体(抑制物)。形成抑制物的风险与疾病的严重程度和因子Ⅷ的暴露相关,前50个暴露日内风险最高,尽管这种风险为少见,但会持续一生。
抑制物形成的临床意义取决于抑制物的滴度,低滴度抑制物临床疗效不佳的风险比高滴度抑制物低。
已有在使用本品后产生抑制物的报告。应通过适当的临床观察或实验室检测,监测患者是否有抑制物形成。如果血浆中的因子Ⅷ活性未达到预期的水平,或在合适的剂量下出血没有得到控制,则应考虑测定因子 Ⅷ 抑制物的滴度。
在存在因子Ⅷ抑制物(尤其是高于 5 BU/mL 的高滴度抑制物)的患者中,因子Ⅷ治疗可能无效,应考虑换用其它治疗方法。这类患者的治疗应由具备治疗血友病和因子Ⅷ抑制物经验的医生指导。
抑制物在 PUP 中很常见,在 PTP 中也有报告。
4. 抗仓鼠蛋白抗体的形成
本品含有微量中国仓鼠卵巢细胞(CHO 细胞)蛋白。在接受本品治疗的患者中,可能对该非人类哺乳动物蛋白产生超敏反应。
5. 实验室检查
- 根据临床指征,通过一期凝固法监测患者血浆的因子Ⅷ活性水平,以确定达到并维持足够的因子Ⅷ水平。
- 根据临床指征,使用因子Ⅷ活性回收率和其它药代动力学参数指导给药剂量。
- 监测因子Ⅷ抑制物的形成。如果血浆中的因子Ⅷ活性未达到预期水平,或在本品推荐剂量下未控制出血,则应监测是否存在因子Ⅷ抑制物,并采用 Bethesda (BU/mL)方法检测抑制物的滴度。
6. 相容性
在缺少非相容性研究的情况下,复溶后的本品不能与其它药品共用同一导管或容器给药。
7. 可追溯性
为提高生物医药产品的可追溯性,应清楚记录所给予药品的名称和批号。
患者可将小瓶上的其中一个剥离式标签贴在日记中,以便记录批号或者用于报告任何副作用。
8. 心血管事件
在已有心血管风险因素的患者中,因子Ⅷ治疗可能增加心血管风险。
9. 导管相关并发症
如需采用中心静脉通路装置(CVAD),应考虑到 CVAD 相关并发症的风险,其中包括局部感染、菌血症和插管部位血栓。
10. 钠含量
复溶之后,每瓶本品含有 1.27 mmol (29 mg)钠,相当于世界卫生组织 (WHO) 建议的成人每日最大摄入量 (RDI)2 g 的 1.5%。根据患者的体重和本品剂量,患者可能需要接受多个小瓶给药。对于低盐饮食的患者,需要考虑这一点。
11. 疗效不足报告
本品临床试验期间和上市后收到的疗效不足报告主要来源于预防使用的患者。疗效不足表现为靶关节出血、新关节出血或新发出血患者的主观感觉。开始使用本品时,建议个体化给药剂量并监测每位患者的因子Ⅷ水平,以确保获得充分的治疗效果。
本品尚未进行动物生殖方面的研究。尚不清楚本品是否对胎儿有害以及是否影响生殖能力。尚无关于因子Ⅷ治疗对分娩影响的研究。尚不清楚本品是否会被分泌至人类乳汁中。
由于血友病 A 在女性中罕有发生,因此缺乏妊娠和哺乳期应用因子Ⅷ产品的经验。妊娠和哺乳期妇女使用本品时须有明确临床指征。
在国外已完成的本品安全性和有效性的开放性研究(n=94)中,17 例年龄为 12 至 16 岁患有重型或重度中间型血友病A(因子Ⅷ:C ≤ 2%)且既往接受过至少 150 天 FⅧ 产品治疗的青少年受试者接受了本品的按需治疗和后续随访。每次注射剂量中位值为 47 IU/kg (最小值-最大值:24 - 74),每例受试者的中位暴露时间为 6 天(最小值-最大值:1 - 26)。
在这 17 例不满 16 岁且接受了至少 1 次本品的受试者中,10 例受试者在研究中出现出血。在接受疗效评估的 10 例受试者中,共按需注射本品治疗了 66 次出血。大部分出血(63/66 或 95.5%)在 1 次或 2 次注射后停止。在 66 次出血中,38 次(57.6%)对初始治疗具有非常好或良好反应,24 次(36.4%)为中等反应,4 次(6.1%)未评估。
在一项针对不满 6 岁患有重度中间型或重型血友病 A(因子Ⅷ:C ≤ 2%)且既往至少接受过 20 天因子Ⅷ 产品的儿童安全性和疗效研究中获得数据。在该研究中,受试者因为出血接受本品按需和随访治疗。每次注射中位值剂量为 28 IU/kg,每例受试者的中位暴露时间为 16 天。
在这 27 例不满 6 岁曾接受了至少 1 剂本品的受试者中,25 例受试者在研究中出现出血。24 例接受疗效评估的受试者共出现了 493 次出血。大部分出血(462/493 或 93.7%)在 1 次或 2 次注射后停止。根据一项四(4)分止血疗效量表评价了受试者的注射效果。在使用本品治疗的 493 次出血中,468 次(94.9%)对初始治疗表现出非常好或良好的反应,22 次(4.5%)为中等反应。
与成人药代动力学参数相比,儿童接受本品后的半衰期较短、分布容积较大、因子 Ⅷ 回收率较低。儿童的清除率(给予每千克体重)大约高 40%。可能需要更高剂量或更频繁的给药来弥补药代动力学参数差异。
在中国儿童患者中进行的上市后 B1831083 研究已完成, 分析了 33 例<6 岁的儿童受试者,以及 36 例 ≥6 至<12岁儿童受试者的数据。 在中国儿童患者中进行的 B1831082 药代动力学研究中,分析了 3 例≥6 至<12 岁儿童受试者的数据,以及 10 例≥ 12 岁受试者的数据。
本品的临床试验尚未纳入≥ 65岁受试者。总体而言,老年患者的剂量选择应个体化。
尚不明确。
尚无重组凝血因子Ⅷ产品使用过量的相关症状的报告。
国外预防治疗和按需治疗的临床试验
在国外研究 310 中,受试者接受本品预防治疗方案,并可根据临床指征接受按需治疗。试验入选了 94 例受试者,并接受了至少一次给药,所有受试者均被纳入意向治疗(ITT)人群中。94 例接受治疗受试者的年龄中位值为 24 岁(均数为27.7 岁,范围为 12~60 岁)。所有受试者的既往暴露日≥150,基线因子Ⅷ活性水平≤ 2 %。89例受试者的治疗暴露日≥50。
在本研究入组的 94 例受试者中,30 例可评估受试者参与了一项随机交叉药代动力学研究。在这些受试者中,因子Ⅷ:C ≤ 1% 的 25 例(25/30)受试者完成了第一次(PK1)和第二次(PK2)药代动力学评估。
在评估安全性和疗效的开放研究阶段,所有 94 例受试者均接受了本品预防性治疗,剂量为30 ± 5 IU/kg, 每周 3 次,并可基于预先确定的标准剂量逐渐递增。试验过程中,有 6 例受试者接受了 7 次剂量递增。94 例受试者中的 43 例受试者(45.7 %)在接受预防性治疗时未报告出血。所有出血事件的年出血率(ABR) 的中位值为 1.9 (均数 3.9,范围0 ~42.1)。
94 例受试者中的 53 例受试者接受了本品的按需治疗,总计治疗了 187 次出血。在这些出血中,有 7 次发生于转换为预防治疗方案前的受试者。180 次出血中的 110 次(61.1 %)发生在末次给药后≤48 小时内,38.9 % (70/180)的出血发生在末次给药后> 48 小时。末次预防给药后≤ 48 小时内的大多数出血(64/110 次出血,58.2 %)是外伤性的。末次预防给药后> 48 小时的出血中有 60 %(42/70 次出血)是自发性的。按需治疗方案由研究者确定。按需治疗的中位剂量为30.6 IU/kg(范围6.4~74.4 IU/kg)。
大多数(173/187; 92.5 %) 出血在 1 次或 2 次注射本品后止血(见表 4)。受试者根据一个 4 分止血效果量表对疗效进行了评定。根据对初始疗效的评价,在本品治疗的 187 次出血中,有 132 次治疗(70.6 %)评定为非常好或好;45 次(24.1 %)评定为一般。5 次 (2.7 %)评定为无反应,5 次(2.7 %)没有评定。
表4. 根据止血所需注射次数分级,针对新发生出血事件疗效的总结
| ------------------注射次数(%)------------------- | ||||||
|---|---|---|---|---|---|---|
| 对首次注射的反应 | 1 | 2 | 3 | 4 | >4 | 总出血次数 |
| 非常好a | 42 (95.5) | 2 (4.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 44 |
| 好b | 69 (78.4) | 16 (18.2) | 3 (3.4) | 0 (0.0) | 0 (0.0) | 88 |
| 一般c | 24 (53.3) | 16 (35.6) | 2 (4.4) | 0 (0.0) | 3 (6.7) | 45 |
| 无反应d | 0 (0.0) | 0 (0.0) | 2 (40.0) | 2 (40.0) | 1 (20.0) | 5 |
| 未评估 | 4 (80.0) | 0 (0.0) | 0 (0.0) | 1 (20.0) | 0 (0.0) | 5e |
| 总计 | 139 (74.3) | 34 (18.2) | 7 (3.7) | 3 (1.6) | 4 (2.1) | 187 |
注:a 很好:一次注射后 8 小时内开始出现明确的疼痛缓解和/或出血体征改善,无需额外注射。
b 良好:一次注射后 8 小时内开始出现明确的疼痛缓解和/或出血体征改善,至少需要一次额外注射才能完全止血。
c 中等:注射后 8 小时后开始出现可能或轻微改善,至少需要一次额外注射才能完全止血。
d 无反应:两次注射之间或注射后 24 小时间隔内没有任何改善,或者病情恶化。
e 包括一次在预防性治疗开始之前,使用另一种已商业化的因子Ⅷ 产品进行的注射。
国外围手术期管理研究
在一项预防手术出血的开放试验中(30 例),入组了 25 例重型或重度中间型(因子 Ⅷ :C ≤ 2%)血友病 A 的 PTPs 接受大手术时应用了本品。
该研究受试者接受的大手术包括11例全膝关节置换术、1 例髋关节置换术、5 例滑膜切除术、1 例左侧尺神经移位松解术、1 例腹疝修补手术/疤痕修补、1 例膝关节镜手术、1 例全膝关节置换术后的膝关节修补和清创术、1 例髋关节置换修复、1 例镫骨置换术、1 例踝关节固定术和 1 例假瘤切除术。表 5 所列为这些患者的止血疗效评价结果。在手术结束时及术后初期,研究者对于所有疗效评估均为非常好或好。所有受试者的术中失血均报告为“正常”或“无”。在这些受试者中,13 例(52%)在术后出现失血。其中 10 例术后失血被评估为“正常”,而另 3 例被评估为“不正常”(1 例由于手术创伤后腹壁动脉出血,1 例由于髋关节置换手术后失血 800 mL,1 例在肘关节滑膜切除术后研究者无法测量失血量)。
| 表 5. 止血疗效总结 | |||
|---|---|---|---|
| 止血疗效评估时间 | 很好a | 良好b | 受试者人数 |
| 手术结束时 | 18 (72%) | 7 (28%) | 25 |
| 首个术后期结束时c | 23 (92%) | 2 (8%) | 25 |
| a 很好:止血效果与非血友病患者接受相似手术后的预期情况相当。 b 良好:止血时间延长,与非血友病患者接受相似手术后的预期情况相比出血有所增加。 c 首个术后期结束日期是出院日期或术后第 6 天,以先到者为准。 | |||
国外儿童患者中的临床疗效
下表中的数据与<12岁患者中进行的本品研究的PUP和PTP数据有关。
表 6. 儿童人群中的用量和疗效结果
| PTP <6岁 | PTP 6-<12岁 | PUP <6岁 | |
|---|---|---|---|
| 每次预防注射的单位体重剂量(IU/kg)a 中位数(min,max) | N=14 36 IU/kg (28, 51) | N=13 32 IU/kg (21, 49) | N=22 46 IU/kg (17,161) |
| 所有受试者的总体ABRb 中位数(min,max) | -- | -- | N=23 3.17 (0.0, 39.5) |
| 基线时接受按需治疗的受试者的总体ABRc 中位数(min,max) | N=5 41.47 (1.6, 50.6) | N=9 25.22 (0.0, 46.6) | -- |
| 基线时接受预防治疗的受试者的总体ABRc 中位数(min,max) | N=13 1.99 (0.0, 11.2) | N=9 5.55 (0.0, 13.0) | -- |
| 每次出血发作时用于出血治疗的单位体重剂量(IU/kg) 中位数(min,max) | N=13 35 IU/kg (28, 86) | N=14 33 IU/kg (17, 229) | N=21 55 IU/kg (11, 221) |
| 通过≤2次注射成功止血的出血事件% | 98.7% | 98.8% | 96.7% |
a 整个研究中规定的本品剂量和频率由研究者根据当地治疗标准决定。
b不要求PUP研究中的受试者遵循定期持续预防治疗;但是,除一例受试者(只有按需(OD)治疗)外,大多数受试者接受定期预防注射。一些受试者开始时接受OD注射,但在参加研究期间换到预防治疗,一些只有零星的预防注射。
c PTP研究中的受试者在基线时报告他们的因子Ⅷ治疗方案(预防或按需),不要求保持这种方案作为参加研究的条件。整个研究中规定的本品剂量和频率由研究者根据当地治疗标准决定。
缩略词:ABR = 年出血率
国内预防和按需治疗的临床试验
在一项已完成的上市后开放试验 B1831083 研究中,共有 73 例受试者根据临床指征接受本品按需治疗。这是一项开放、单臂、多中心、前瞻性、注册后实用性研究,在中国的血友病治疗研究中心内进行。按照当地医疗标准监测受试者。该研究在按需治疗人群中分析了本品在不同血友病 A患 者人群(尤其是<6 岁儿童患者、≥6 至<12 岁儿童患者、既往未接受过治疗的患者(PUP)、预防性治疗患者以及重度(因子Ⅷ活性<1%)的患者)中的疗效。
在总人群中,为治疗每次新出血事件的平均(标准差[SD])本品注射次数为 1.6 (0.94)。在<6 岁的受试者中,为治疗每次新出血事件的平均(SD)本品注射次数为 1.7 (1.04)。在≥6 到<12 的受试者中,为治疗每次新出血事件的平均(SD)本品注射次数为 1.5 (0.89)。在 PUP 中,为治疗每次新出血事件的平均(SD)本品注射次数为 1.6 (1.00)。在重型受试者中,为治疗每次新出血事件的平均(SD)本品注射次数为 1.8 (1.03)。
总体而言 1015/1610(63.0%)次出血通过 1 次注射解决,352 (21.9%)次出血通过 2 次注射解决,139 (8.6%)次出血通过 3 次注射解决,96 (6.0%)次出血通过 4 次注射解决,8 (0.5%)次出血通过大于 4 次注射解决。总体而言,大多数出血治疗的注射评分为“非常好”(46.9%)或“好”(40.0%)(表 7)。
表7. 按需治疗出血缓解效果评估总结
| 止血疗效,n (%) | 首次注射 (N = 1610) | 后续注射 (N = 958) | 所有注射 (N = 2568) |
|---|---|---|---|
| 非常好 | 741(46.0) | 463(48.3) | 1204(46.9) |
| 好 | 633(39.3) | 395(41.2) | 1028(40.0) |
| 一般 | 229(14.2) | 98(10.2) | 327 (12.7) |
| 无反应 | 5(0.3) | 2(0.2) | 7 (0.3) |
| 未记录 | 2(0.1) | 0 | 2(0.1) |
缩写:n = 观察数量;N =因任意出血接受治疗的注射数量。
有 14 例受试者进行了手术预防注射。在手术当天,止血效果由研究者评分为“非常好”(71.4%)或“好”(28.6%),术后评分为“非常好”(75.5%)和“好”(24.5%)。详见下表 8。
表 8. 手术预防治疗的止血疗效总结
| 止血疗效,n (%) | 手术日 (N = 14) | 术后 (N = 14) |
|---|---|---|
| 非常好 | 10 (71.4) | 148 (75.5) |
| 好 | 4 (28.6) | 48 (24.5) |
| 一般 | 0 | 0 |
| 无反应 | 0 | 0 |
| 总计 | 14 | 196 |
缩写:n = 观察数量;N = 在手术预防性治疗期间进行止血疗效评估的受试者数量。
药理作用
在内源性凝血过程中,活化的因子Ⅷ是活化因子 IX 的辅因子,可加快因子 X 转化为活化的因子 X。活化的因子 X 可将凝血酶原转化为凝血酶,凝血酶将纤维蛋白原转化为纤维蛋白,形成凝块。
本品为不含 B 结构域的重组人凝血因子Ⅷ,其功能特点与内源性因子Ⅷ相当。血友病 A 患者缺乏因子Ⅷ。本品可暂时性取代有效凝血所必须的因子Ⅷ,本品给药可升高因子Ⅷ的血浆水平,从而使这些患者的凝血缺陷异常得到暂时纠正。
血友病患者活化部分凝血活酶时间(aPTT)延长。aPTT 的测定是一种常用的凝血因子Ⅷ体外生物学活性试验,本品在有效的治疗期后可使 aPTT 正常化。
毒理研究
一般毒理学试验中,与本品相关的毒性主要与抗因子Ⅷ中和抗体产生有关,该抗体在给予高剂量(约 735IU/kg/天)的非人类灵长类动物中重复给药 15 天后首次检测到。
国外的药代动力学试验
表 9总结了年龄为 12 至 60 岁的 30 例经治成年患者接受单次注射 50 IU/kg 本品后 的药代动力学参数。在本试验中,25 例受试者后来接受剂量为 50 IU/kg的本品单次注射,之后随访 6 个月。基线与第 6 个月比较,药代动力学参数相似,表明本品的药代动力学特点不存在时间依赖性变化。
在另一项研究中,在 30 例年满 12 岁的血友病A受试者中,8 例接受择期大手术的受试者接受了单次注射 50 IU/kg 本品。表 9 还总结了这些受试者的药代动力学参数。
| 表 9. 经治血友病 A 患者接受单次注射 50 IU/kg 本品后的药代动力学参数(平均 ± SD) | |||
|---|---|---|---|
| 参数 | 首次访视 (n = 30) | 第 6 个月 (n = 25) | 手术前 (n=8) |
| Cmax(IU/mL) AUC∞(IU•hr/mL) t1/2(hr) CL(mL/hr/kg) Vss(mL/kg) 回收率[(IU/dL)/(IU/kg)] | 1.08 ± 0.22 13.5 ± 5.6 11.2 ± 5.0 4.51 ± 2.23 66.1 ± 33.0 2.15 ± 0.44 | 1.24 ± 0.42 15.0 ± 7.5 11.8 ± 6.2* 4.04 ± 1.87 67.4 ± 32.6 2.47 ± 0.84 | 1.08 ± 0.24 16.0 ± 5.2 16.7 ± 5.4 3.48 ± 1.25 69.0 ± 20.1 2.17 ± 0.47 |
| 缩写:AUC∞ = 从零至无穷大的血浆浓度时间曲线下面积;Cmax = 峰值浓度;t1/2 = 血浆消除半衰期;CL = 清除率;n = 受试者数量;SD = 标准差;Vss = 稳态分布容积。 *一例受试者由于无法界定终末期而被排除在计算之外。 | |||
表 10 显示了 9 名儿童接受单剂量 50 IU/kg 本品后的药代动力学参数;其中 4 名年龄为 14 或 15 岁,他们也被纳入上述成人总结中,另外 5 名儿童年龄为 3.7–5.8 岁。与成人相比,儿童的半衰期较短,清除率(基于每千克体重)大约高 40%。
| 表 10. 经治血友病 A 儿童患者接受单次注射 50 IU/kg 本品后的药代动力学参数(平均 ± SD) | ||
|---|---|---|
| 参数 | 幼儿(n=5) | 青少年(n=4) |
| 年龄(最小值-最大值,岁) Cmax(IU/mL) AUC∞(IU•hr/mL) t1/2(hr) CL(mL/hr/kg) Vss(mL/kg) 回收率[(IU/dL)/(IU/kg)] | 3.7–5.8 0.78 ± 0.34 12.2 ± 6.50 8.3 ± 2.7 6.29 ± 4.87 66.9 ± 55.6 1.52 ± 0.69 | 14–15 0.97 ± 0.21 8.5 ± 4.0 6.9 ± 2.4 6.62 ± 2.16 67.1 ± 13.6 1.95 ± 0.41 |
| 缩写:AUC∞ = 从零至无穷大的血浆浓度时间曲线下面积;Cmax = 峰值浓度;t1/2 = 血浆消除半衰期;CL = 清除率;n = 受试者数量;SD = 标准差;Vss = 稳态分布容积。 | ||
在 19 例PUP的研究中,17 例 28 天至 2 岁以下儿童在研究开始时回收率为 1.32 ± 0.65 (IU/dL)/(IU/kg),2 例 2-<6 岁儿童为 1.7 和 1.8 (IU/dL)/(IU/kg)。除了检测到抑制物的病例,不同时间的平均回收率稳定(2 年期间6 次访视),个体值在 0(存在抑制物)至 2.7 (IU/dL)/(IU/kg)范围内。
下表显示在 37 例儿童 PTP 的研究中,50 IU/kg给药一次后观察到的本品的药代动力学参数。
| 表11. 儿童 PTP 中 50 IU/kg单次给药后因子Ⅷ药代动力学参数(平均±SD) | ||
|---|---|---|
| PK 参数 | 受试者人数 | 平均值a ± SD |
| 回收率,(IU/dL)/(IU/kg) 年龄<6 岁 年龄 6-<12 岁 | 17 19 | 1.7 ± 0.4 2.1 ± 0.8 |
| Cmax, IU/mLb | 19 | 0.9 (45) |
| AUCinf, IU∙h/mLb | 14 | 9.9 (41) |
| t½, hb | 14 | 9.1 ± 1.9 |
| CL, mL/h/kgb | 14 | 4.4 (30) |
| Vss, mL/kgb | 14 | 56.4 (15) |
a 除了增量回收率和t½显示算数平均值±SD,所有其他值均为几何平均值(几何CV%)。
b 只有 6-<12 岁的患者。
缩略词:Cmax = 观察到的最高血浆浓度;CV = 变异系数;AUCinf = 从 0 时外推到无限时间的血浆浓度-时间曲线下面积;t½ = 终末消除半衰期;CL = 清除率;Vss = 稳态分布容积。
儿科人群(12~16 岁)
在国外进行的 8 例既往接受过治疗的 12~16 岁患者中进行了本品药代动力学研究。在这些儿童患者中,药代动力学参数与成人接受 50 IU/kg一次给药后的结果相似。7例患者平均(± SD) Cmax和 AUC∞分别为 1.09 ± 0.21 IU/mL和 11.5 ± 5.2 IU•h/mL,平均清除率和血浆半衰期分别为 5.23 ± 2.36 mL/h/kg 和 8.03 ± 2.44 小时(范围 3.52~10.6 小时),平均K值和体内回收率分别为 2.18 ± 0.41 (IU/dL) / (IU/kg) 和 112 ± 23%。
中国的药代动力学试验
B1831082 是一项在中国男性血友病 A 受试者中进行的多中心、开放性、单剂量研究。本研究分析了 3 例≥6 至<12 岁亚组受试者的数据,以及 10 例 ≥12 岁亚组受试者的数据。受试者在给予本品的第一天之前至少三天(至少 72 小时)内,必须未接受过任何凝血因子Ⅷ制剂的治疗,包括任捷。
受试者参与研究最长达 33 天。筛选期和第-1日之间的 28 天、第 0 日、以及第 1 日给药后的4天。第 1 日,在 10 分钟内静脉注射单剂量 50IU/kg 本品。
在针对中国受试者进行的本研究中,观察到的 PK 参数与其他研究的结果类似。本研究中单剂量静脉注射本品 50 IU/kg是安全的、且耐受性良好。与较年长的儿童和成年人相比,6-12 岁的儿童暴露量较低(AUCinf, AUClast和Cmax),分布容积较高,回收率较低,CL较高,t½较短。表 12描述性总结了 PK 参数。
表 12. 血浆因子Ⅷ活性药代动力学参数值的描述性总结
| 参数,单位 | 不同年龄组参数的统计学小结a本品 50 IU/kg | ||
|---|---|---|---|
| ≥6 至<12岁 | ≥12岁 | 所有受试者 | |
| N AUCinf, IU•hr/mL AUClast, IU•hr/mL Cmax, IU/mL Tmax, hr kel, 1/hr t½, hr MRT, hr CL, mL/hr/kg Vss, mL/kg 增量回收率 (IU/dL)/(IU/kg) | 3 7.931 (50) 7.702 (51) 0.8952 (18) 0.250 (0.250 – 0.500) 0.09844 (24) 7.173 ±1.77 10.09 (27) 6.653 (30) 67.18 (10) 1.695 (5) | 10 18.48 (41) 17.51 (40) 1.235 (47) 0.500 (0.250 – 3.00) 0.05215 (28) 13.76 ±3.77 18.92 (26) 2.669 (38) 50.53 (30) 2.498 (47) | 13 15.21 (58) 14.49 (57) 1.147 (44) 0.500 (0.250 – 3.00) 0.06039 (39) 12.24 ±4.42 16.37 (38) 3.295 (56) 53.96 (29) 2.284 (44) |
药代动力学参数定义见表 9。
缩写: %CV = 百分比变异系数,因子Ⅷ = 凝血因子Ⅷ, hr = 小时, IU = 国际单位, N = 纳入统计小结的受试者人数,SD = 标准差,Mean residence time (MRT) =平均滞留时间。
a. 均为几何学平均值 (%CV) ,除Tmax的中位数(范围),t½. 平均 ± SD为算术平均值。
于 2~8 °C 避光保存和运输。禁止冷冻。
本品包装内含:
1 瓶注射用重组人凝血因子Ⅷ冻干粉;
1 支预装 4 ml 0.9 % 氯化钠稀释液的注射器及推杆;
1 个接合器;
1 支无菌静脉输液针。
36 个月。
进口药品注册标准 JS20220040
250 IU/瓶:国药准字 SJ20150003
500 IU/瓶:国药准字 SJ20150005
1000 IU/瓶:国药准字 SJ20150006
2000 IU/瓶:国药准字 SJ20150004
名 称:Pfizer Europe MA EEIG
注册地址:Boulevard de la Plaine 17, 1050 Bruxelles, Belgium
企业名称:Wyeth Farma S. A.
生产地址:Autovia del Norte A-1 Km 23, Desvio Algete Km 1, 28700 San Sebastian de los Reyes, Madrid, 西班牙。
邮政编码:28700
名 称:辉瑞投资有限公司
地 址:上海市南京西路1168号中信泰富广场36层
联系方式:400 910 0055
2012年08月16日
2014年08月10日;2015年01月20日;2016年09月21日;2017年03月15日;2018年06月28日;2019年05月24日;2020年09月01日;2021年05月14日;2022年09月02日;2022年11月15日;2025年07月01日;2026年01月01日



