
简要介绍
前列腺癌
1. Eur Urol:前列腺特异性膜抗原正电子发射断层显像在前列腺癌确诊治疗前诊断及初始分期中作用的全面系统评价与荟萃分析
2. Lancet Oncol:一线他拉唑帕利联合恩扎卢胺对比安慰剂联合恩扎卢胺治疗转移性去势抵抗性前列腺癌:来自随机、双盲、安慰剂对照3期TALAPRO-2试验的患者报告结局
3. Lancet Oncol:一线他拉唑帕利联合恩扎卢胺对比安慰剂联合恩扎卢胺治疗同源重组修复基因突变的转移性去势抵抗性前列腺癌患者:来自随机、双盲、安慰剂对照3期TALAPRO-2试验的患者报告结局
4. J Clin Oncol:帕博利珠单抗联合多西他赛对比多西他赛治疗既往接受过治疗的转移性去势抵抗性前列腺癌:随机、双盲、Ⅲ期KEYNOTE-921试验
5. J Clin Oncol:FOR46(FG-3246),一种靶向CD46的免疫调节抗体-药物偶联物,在转移性去势抵抗性前列腺癌患者中的I期首次人体研究
6. Lancet Oncol:前列腺癌的低分割放疗(HYDRA):MARCAP联盟中随机试验的个体患者数据荟萃分析
7. JAMA Oncol:前列腺癌根治性前列腺切除术后前列腺特异性抗原持续存在与死亡风险
肾癌
8. Nat Cancer:局部晚期非转移性肾透明细胞癌的新辅助卡博替尼治疗:一项2期临床试验
9. Lancet Oncol:晚期肾细胞癌患者使用贝组替凡对比依维莫司的健康相关生活质量(LITESPARK-005):来自一项随机、开放标签、3期试验的患者报告结局
10. Ann Oncol:伊匹木单抗/纳武利尤单抗对比标准治疗方案用于非透明细胞肾细胞癌的前瞻性随机II期试验 ——SUNNIFORECAST 试验结果
前列腺癌
1.前列腺特异性膜抗原正电子发射断层显像在前列腺癌确诊治疗前诊断及初始分期中作用的全面系统评价与荟萃分析
发表期刊:Eur Urol
背景与目的:前列腺特异性膜抗原(PSMA)正电子发射断层显像(PET)在前列腺癌(PCa)患者的诊断和初始分期中已确立了一定作用,但目前仍缺乏关于其实际诊断和分期价值的最新总结性证据。我们旨在收集并分析已发表的研究,这些研究报告了 PSMA PET 在诊断具有临床意义的前列腺癌(csPCa)以及在确诊治疗前初始分期时检测远处转移方面的准确性。
方法:我们对文献进行了系统综述,检索了 PubMed/MEDLINE、考克兰图书馆的 CENTRAL、EMBASE 和 Scopus 数据库,检索从数据库建立至 2024 年 4 月的所有资料。评估了两个主要结局:其一,在以患者为单位的层面上,评估 PSMA PET 检测前列腺内具有临床意义的前列腺癌的敏感性、特异性、阳性预测值(PPV)和阴性预测值(NPV);其二,在确诊治疗前,评估初始分期时转移性疾病的阳性率。作为次要结局,在以患者为单位的层面上,对那些以盆腔淋巴结清扫术(PLND)作为参考标准的研究进行分析,检验 PSMA PET 检测淋巴结侵犯(LNI)的诊断准确性。使用随机效应模型汇总阳性率和检测率。预先计划的亚组分析检验了 PSMA PET 在不同研究队列中的诊断准确性。评估了在不同 csPCa 和 LNI 患病率情况下,阳性预测值和阴性预测值的变化情况。
关键发现与局限性:在前列腺内诊断和分期的定量综合分析中,总共纳入了 12 项和 99 项研究,分别涉及 1533 例和 18649 例受试者。对于前列腺内疾病,PSMA PET 检测 csPCa 的敏感性、特异性、阳性预测值和阴性预测值分别为 82%(95%置信区间[CI]73–90%)、67%(95% CI: 46– 85%)、77%(95% CI: 63–88%)和 73%(95% CI: 56–87%)。在双变量分析中,通过汇总受试者工作特征曲线得出的曲线下面积来估计,PSMA PET 的诊断准确性为 84%,当与磁共振成像(MRI)结合时,该值可提高至 88%。在分期层面,23% 的患者在前列腺外 PSMA PET 结果呈阳性,高危亚组(31%)和中危亚组(12%)之间的阳性率存在显著差异。当以盆腔淋巴结清扫术作为参考标准时( 51 项研究,7713 例患者),PSMA PET 的敏感性、特异性、阳性预测值和阴性预测值分别为 54%、94%、77% 和 86%。随着 csPCa 和 LNI 患病率的升高,观察到阳性预测值有类似的升高,而阴性预测值则下降。
结论与临床意义:当前更新的系统评价和荟萃分析为 PSMA PET 在前列腺癌中的诊断和分期准确性提供了最新证据。我们发现 PSMA PET 在鉴别 csPCa 方面具有良好的准确性,尤其是与 MRI 结合使用时,但仅靠阴性预测值不足以省略活检步骤。关于分期,PSMA PET 不能单独用于确定是否需要进行淋巴结清扫术(LND),而应在预测工具中结合其他临床信息。因此,进一步的研究应开发并验证纳入 PSMA PET 的模型,以便可靠地为活检或淋巴结清扫术提供依据。
2.一线他拉唑帕利联合恩扎卢胺对比安慰剂联合恩扎卢胺治疗转移性去势抵抗性前列腺癌:来自随机、双盲、安慰剂对照3期TALAPRO-2试验的患者报告结局
发表期刊:Lancet Oncol
背景:转移性去势抵抗性前列腺癌患者预后较差,这凸显了对新型治疗策略的需求。在 3 期 TALAPRO-2 研究中,与安慰剂联合恩扎卢胺相比,一线他拉唑帕利联合恩扎卢胺显著改善了转移性去势抵抗性前列腺癌男性患者的影像学无进展生存期。我们旨在评估 TALAPRO-2 研究中所有受试者队列的患者报告结局,该队列包括同源重组修复(HRR)基因有无改变的患者。
方法:TALAPRO-2 是一项随机、双盲、安慰剂对照的 3 期试验,在全球 26 个国家的 223 家医院、癌症中心和医疗中心开展。符合条件的受试者为年龄在 18 岁及以上(在日本为≥20岁)的男性患者,这些患者正在接受持续的雄激素剥夺治疗,患有无症状或轻度症状的转移性去势抵抗性前列腺癌,东部肿瘤协作组体能状态评分为 0或1,且此前未接受过针对去势抵抗性前列腺癌或转移性去势抵抗性前列腺癌的延长生命的全身治疗。患者使用集中式交互式网络响应系统,按照 4 的随机区组大小以 1:1 的比例随机分配,分别接受口服他拉唑帕利 0.5 mg 每日一次或安慰剂,同时均联合口服恩扎卢胺 160 mg 每日一次。资助方、患者和研究人员对他拉唑帕利或安慰剂的分配情况不知情;恩扎卢胺为开放标签。分层因素为 HRR 基因改变状态(缺陷型与非缺陷型或未知型)以及在去势敏感阶段是否曾接受过多西他赛或阿比特龙或两者皆用的治疗(是与否)。主要终点是由盲态独立中心审查评估的影像学无进展生存期,此前已有相关报道。患者报告结局作为次要终点在患者报告结局人群中进行评估,该人群由意向性治疗人群中基线时有患者报告结局评估且至少有一次基线后患者报告结局评估的患者组成。患者报告结局包括患者报告的疼痛症状(根据简明疼痛量表简表[BPI-SF])与基线相比的平均变化;总体健康状况/生活质量(GHS/QoL)、总体癌症及前列腺癌特异性功能和症状(根据欧洲癌症研究与治疗组织[EORTC]核心生活质量问卷[QLQ-C30]和前列腺癌生活质量问卷[QLQ-PR25]);以及一般健康状况(根据EQ-5D-5L)。患者报告的疼痛症状(根据BPI-SF)恶化时间,以及患者报告的总体健康状况/生活质量(根据EORTC QLQ-C30)和前列腺癌特异性泌尿症状(根据EORTC-QLQ-PR25)明确恶化时间是其他次要终点。本研究已在 ClinicalTrials.gov 注册,编号为 NCT03395197,目前仍在进行中。
结果:在 2019 年 1 月 7 日至 2020 年 9 月 17 日期间,805 例患者入组并被随机分配接受治疗,无论HRR基因改变状态如何。395 例被分配至他拉唑帕利联合恩扎卢胺组和398例被分配至安慰剂联合恩扎卢胺组的患者被纳入患者报告结局人群。他拉唑帕利联合恩扎卢胺组的中位随访时间为 28.0 个月(四分位距23.9-31.7),安慰剂联合恩扎卢胺组为 26.8 个月(23.4-30.6)。如图 1,与安慰剂联合恩扎卢胺相比,他拉唑帕利联合恩扎卢胺组的总体健康状况/生活质量明确恶化时间更长(中位 30.8 个月[95% CI: 27.0-无法估计]对比25.0个月[95% CI: 22.9-30.7];风险比HR=0.78[95% CI: 0.62-0.99];双侧 P=0.038)。他拉唑帕利联合恩扎卢胺组的泌尿症状明确恶化的中位时间无法估计(95% CI: 无法估计-无法估计),安慰剂联合恩扎卢胺组为 35.9 个月(95% CI: 32.3-无法估计)(HR=0.76[95% CI: 0.54-1.06];双侧 P=0.11)。治疗组之间在总体健康状况/生活质量、症状和功能量表方面与基线相比的平均变化未观察到具有临床意义的差异(≥10分)。两组之间在根据 BPI-SF 测量的疼痛恶化时间方面未观察到差异(HR=0.98[95% CI: 0.69-1.40];双侧 P=0.93),平均疼痛评分方面(治疗组之间过去 24 小时内最严重疼痛值的估计平均差值为-0.1[95% CI: -0.3-0.1];双侧 P=0.27),或根据 EQ-5D-5L 测量的一般健康状况方面(估计平均差值0.0[95% CI: 0.0-0.0];双侧 P=0.37)也均未观察到差异。
结论:与安慰剂联合恩扎卢胺相比,他拉唑帕利联合恩扎卢胺延长了总体健康状况/生活质量的明确恶化时间。结合临床疗效和安全性数据,这些结果为 TALAPRO-2 研究中转移性去势抵抗性前列腺癌患者使用他拉唑帕利联合恩扎卢胺的风险-获益评估提供了参考。
图1.两组患者根据EORTC QLQ-C30报告的GHS/QoL最终恶化的时间

3.一线他拉唑帕利联合恩扎卢胺对比安慰剂联合恩扎卢胺治疗同源重组修复基因突变的转移性去势抵抗性前列腺癌患者:来自随机、双盲、安慰剂对照3期TALAPRO-2试验的患者报告结局
发表期刊:Lancet Oncol
背景:在 3 期 TALAPRO-2 试验中,对于携带同源重组修复(HRR)相关基因突变的转移性去势抵抗性前列腺癌男性患者,他拉唑帕利联合恩扎卢胺相较于安慰剂联合恩扎卢胺,显著改善了影像学无进展生存期。我们旨在评估 TALAPRO-2 试验中 HRR 缺陷型转移性去势抵抗性前列腺癌患者的患者报告结局。
方法:TALAPRO-2 是一项随机、双盲、安慰剂对照的 3 期试验,在全球 26 个国家的 223 家医院、癌症中心和医疗中心开展。符合条件的受试者为年龄 18 岁及以上(在日本为≥20岁)的男性患者,这些患者正在接受持续的雄激素剥夺治疗,患有无症状或轻度症状的转移性去势抵抗性前列腺癌,东部肿瘤协作组体能状态评分为 0或1,且此前未接受过针对去势抵抗性前列腺癌或转移性去势抵抗性前列腺癌的延长生命的全身治疗。对于HRR基因改变的患者,使用集中式交互式网络响应系统,按照4的随机区组大小以 1:1 的比例随机分配,分别接受口服他拉唑帕利 0.5 mg 每日一次或安慰剂,同时均联合口服恩扎卢胺 160 mg 每日一次,并根据在去势敏感阶段是否曾接受过第二代雄激素受体通路抑制剂(阿比特龙或奥替瑞林)或多西他赛治疗(是与否)进行分层。申办方、患者和研究人员对他拉唑帕利或安慰剂的分配情况不知情;恩扎卢胺为开放标签。主要终点是由盲态独立中心审查评估的影像学无进展生存期,此前已有相关报道。患者报告结局作为次要结局在患者报告结局人群中进行评估,该人群由意向性治疗人群中基线时有患者报告结局评估且至少有一次基线后患者报告结局评估的患者组成。患者报告结局包括根据欧洲癌症研究与治疗组织(EORTC)核心生活质量问卷(QLQ-C30)评估的总体健康状况/生活质量(GHS/QoL)明确恶化时间、根据 EORTC 前列腺癌生活质量问卷(QLQ-PR25)评估的前列腺癌特异性泌尿症状明确恶化时间,以及根据简明疼痛量表简表(BPI-SF)评估的疼痛症状恶化时间。根据 EORTC QLQ-C30 和 QLQ-PR25 评估的 GHS/QoL、总体癌症及前列腺癌特异性功能和症状与基线相比的平均变化,根据BPI-SF评估的疼痛症状与基线相比的平均变化,以及根据 EQ-5D-5L 评估的一般健康状况与基线相比的平均变化,也均属于患者报告的次要结局。本研究已在 ClinicalTrials.gov 注册,编号为 NCT03395197,目前仍在进行中。
结果:在 2018 年 12 月 18 日至 2022 年 1 月 20 日期间,399 例HRR缺陷型转移性去势抵抗性前列腺癌患者入组并被随机分配,其中 197 例被分配至他拉唑帕利联合恩扎卢胺组和 197 例被分配至安慰剂联合恩扎卢胺组的患者被纳入患者报告结局人群。他拉唑帕利联合恩扎卢胺组的中位随访时间为 22.2 个月(四分位距13.8-27.7),安慰剂联合恩扎卢胺组为 20.2 个月(13.5-26.6)。如图 2,他拉唑帕利联合恩扎卢胺组的 GHS/QoL 明确恶化的中位时间(27.1个月[95% CI: 21.2-无法估计])长于安慰剂联合恩扎卢胺组( 19.3 个月[95% CI: 16.6-23.0];风险比HR=0.69[95% CI:0.49-0.97];双侧 P=0.032)。他拉唑帕利联合恩扎卢胺组的泌尿症状明确恶化的中位时间(无法估计[95% CI: 32.2-无法估计])也长于安慰剂联合恩扎卢胺组(30.2 个月[95% CI: 24.6-无法估计];HR=0.56[0.34-0.93];双侧 P=0.022)。两个治疗组的疼痛症状恶化的中位时间均无法估计(HR=0.58[0.33-1.01];双侧 P=0.051)。过去 24 小时内最严重疼痛(BPI-SF,问题三)和一般健康状况(EQ-5D-5L)与基线相比的变化情况,他拉唑帕利联合恩扎卢胺组也优于安慰剂联合恩扎卢胺组,尽管这些差异不具有临床意义。根据 EOR TCQLQ-C30 评估,两组之间在 GHS/QoL、功能和症状方面与基线相比的平均变化差异未达到 10 分或以上的临床意义阈值,不过在身体、情感和认知功能以及疼痛方面,他拉唑帕利联合恩扎卢胺组更具优势。同样,根据 EORTC QLQ-PR25 评估,在泌尿和肠道症状方面与基线相比的平均变化差异倾向于他拉唑帕利联合恩扎卢胺组,但不具有临床意义。
结论:与安慰剂联合恩扎卢胺相比,他拉唑帕利联合恩扎卢胺在 HRR 缺陷型转移性去势抵抗性前列腺癌患者中表现出对 GHS/QoL、泌尿症状以及其他功能和症状量表明确恶化的延迟作用,这为这些患者的临床决策提供了有价值的参考。
图2.两组患者根据EORTC QLQ-C30报告的GHS/QoL最终恶化的时间

4.帕博利珠单抗联合多西他赛对比多西他赛治疗既往接受过治疗的转移性去势抵抗性前列腺癌:随机、双盲、Ⅲ期KEYNOTE-921试验
发表期刊:J Clin Oncol
目的:经第二代雄激素受体通路抑制剂(ARPI)治疗后的转移性去势抵抗性前列腺癌(mCRPC)的标准治疗方案仍为多西他赛。这项随机、双盲、Ⅲ 期 KEYNOTE-921 试验(Clinicaltrials.gov 注册号:NCT03834506)评估了帕博利珠单抗或安慰剂联合多西他赛用于既往接受过治疗的 mCRPC 患者的疗效与安全性。
方法:患有 mCRPC 且在雄激素剥夺治疗和一种 ARPI 治疗后出现疾病进展的成年患者,按 1:1 的比例随机分配,分别接受帕博利珠单抗或安慰剂联合多西他赛,并同时使用泼尼松。双主要终点为根据前列腺癌工作组 3 修改版的实体瘤疗效评价标准 1.1(RECIST 1.1),由盲态独立中心审查评估的影像学无进展生存期(rPFS)和总生存期(OS)。安全性为次要终点。
结果:在 2019 年 5 月 30 日至 2021 年 6 月 17日 期间,515 例受试者被随机分配至帕博利珠单抗联合多西他赛组,515 例被分配至安慰剂联合多西他赛组。在最终分析(FA)时,从随机分组到数据截止日期(2022 年 6 月 20 日)的中位时间为 22.7 个月(范围:12.1-36.7 个月)。如图 3,在首次中期分析时(数据截止日期:2021 年 9 月 27 日),帕博利珠单抗联合多西他赛组的中位 rPFS 为 8.6 个月(95% CI: 8.3-10.2),而安慰剂联合多西他赛组为 8.3 个月(95% CI:,8.2-8.5)(HR=0.85[95% CI:,0.71-1.01;P=0.03)。如图 4,在最终分析时,两组的中位OS分别为19.6 个月(95% CI: 18.2-20.9)和 19.0 个月(95% CI: 17.9-20.9)(HR=0.92[95% CI: 0.78-1.09;P=0.17)。接受帕博利珠单抗联合多西他赛治疗的受试者中,43.2%发生了 ≥3级的治疗相关不良事件,而接受安慰剂联合多西他赛治疗的受试者中这一比例为 36.6%。分别有 2 例和 7 例受试者因治疗相关不良事件死亡。肺炎是最常见的免疫介导不良事件(7.0%对比3.1%)。
结论:对于既往接受过治疗的 mCRPC 患者,在多西他赛基础上加用帕博利珠单抗并未显著改善疗效结局。目前的标准治疗方案保持不变。
图3.意向治疗人群的影像学无进展生存期

图4.意向治疗人群的总生存期

5.FOR46(FG-3246),一种靶向CD46的免疫调节抗体-药物偶联物,在转移性去势抵抗性前列腺癌患者中的I期首次人体研究
发表期刊:J Clin Oncol
目的:FOR46 是一种与单甲基奥瑞他汀E偶联的全人源抗体,靶向 CD46 的肿瘤选择性表位,而 CD46 在转移性去势抵抗性前列腺癌(mCRPC)中存在过表达情况。FOR46 在恩扎卢胺耐药的去势抵抗性前列腺癌(CRPC)模型中显示出强大的非临床活性。
患者与方法:这是一项针对在接受≥一种雄激素信号抑制剂治疗后病情进展的 mCRPC 患者的 I 期首次人体剂量递增/扩展研究(ClinicalTrials.gov 注册号:NCT03575819)。FOR46 的起始剂量为每3周静脉注射 0.1 mg/kg。主要目的是确定最大耐受剂量(MTD)。采用全血质谱流式细胞术(飞行时间流式细胞术)来测定经中心病理检查的CRPC组织中的外周免疫反应和 CD46 表达情况。
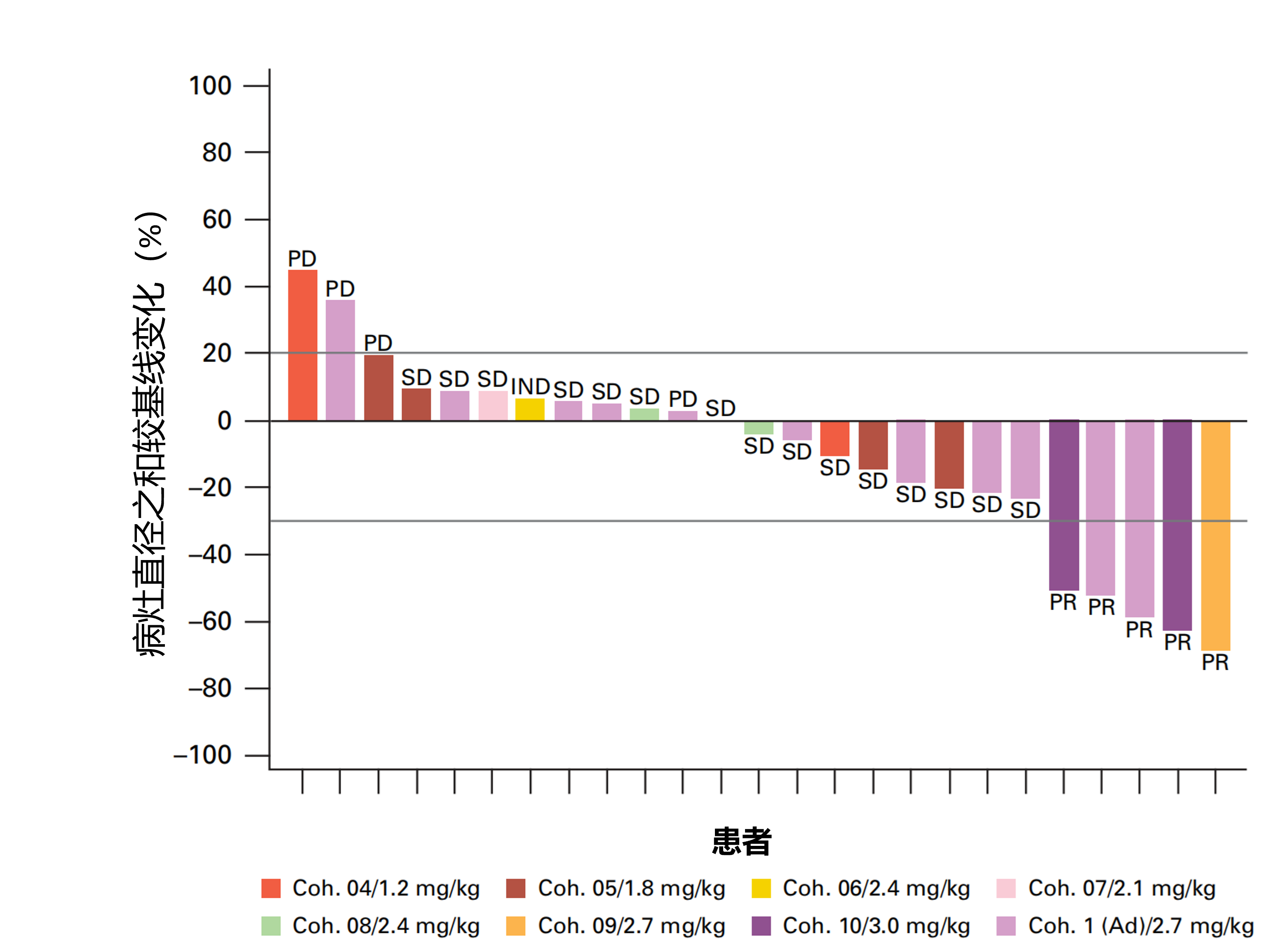
结果:共纳入了 56 例患者。剂量限制性毒性包括中性粒细胞减少(n=4)、发热性中性粒细胞减少(n=1)和疲劳(n=1)。使用校正体重计算得出的最大耐受剂量为 2.7 mg/kg。在所有剂量水平中,最常见的 ≥3级不良事件为中性粒细胞减少(59%)、白细胞减少(27%)、淋巴细胞减少(7%)、贫血(7%)和疲劳(5%)。观察到 1 例 3 级发热性中性粒细胞减少事件。未发生与治疗相关的死亡病例。在疗效可评估亚组(起始剂量≥1.2 mg/kg的腺癌患者,n=40)中,中位影像学无进展生存期为 8.7 个月(范围:0.1-33.9 个月)。39 例可评估患者中有 14 例(36%)达到了前列腺特异性抗原(PSA)下降 50%的反应(PSA50反应)。经确认的客观缓解率为 20%( 25 例根据实体瘤疗效评价标准[RECIST]可评估的患者中有 5 例,见图 5)。中位缓解持续时间为 7.5 个月。有反应的患者在治疗期间循环效应性 CD8+T 细胞的频率显著更高。
结论:FOR46 显示出令人鼓舞的初步临床活性,且安全性可控。靶向 CD46 引发了与临床结局相关的免疫启动效应。
图5.39例可评估患者与基线相比病灶大小变化的百分比
6.前列腺癌的低分割放疗(HYDRA):MARCAP联盟中随机试验的个体患者数据荟萃分析
发表期刊:Lancet Oncol
背景:在针对前列腺癌的研究中,比较中度低分割放疗(MHFRT)与常规分割放疗(CFRT)的试验,在研究目的(非劣效性与优效性)以及中度低分割放疗的剂量方面存在很大差异。我们对等剂量中度低分割放疗和剂量递增的中度低分割放疗的疗效和毒性特征进行比较。
方法:这是一项个体患者数据荟萃分析,旨在找出已发表了关于疗效和晚期毒性的个体患者层面数据的、比较常规分割放疗与中度低分割放疗的随机3期试验。最初于 2023 年 12 月 15 日使用 MEDLINE、Embase、试验注册库、the Web of Science,、Scopus 以及相关会议记录进行了系统的文献检索,并于 2025 年 1 月 8 日重新进行了检索。未发表疗效数据、未发表晚期毒性数据,或者在常规分割放疗组中未使用现代剂量放疗(以2戈瑞当量计算≥70戈瑞)的试验均被排除在外。研究调查人员向MARCAP提供了个体患者数据。设计了三项独立的荟萃分析,以比较接受常规分割放疗与中度低分割放疗的患者之间的疗效(主要终点是无进展生存期)、医生评分的晚期毒性(共同主要终点是 2级或更高级别的晚期泌尿生殖系统毒性和 2级或更高级别的晚期胃肠道毒性效应)以及患者报告结局(共同主要终点是患者报告的泌尿或肠道生活质量出现具有临床意义的下降)。
结果:我们确定了 1696 条记录以供审查。七项比较中度低分割放疗与常规分割放疗的 3 期试验符合纳入我们分析的标准。从这七项研究中获取了个体患者数据(来自三项比较常规分割放疗与等剂量中度低分割放疗的试验的 3454 例患者,以及来自四项比较常规分割放疗与剂量递增的中度低分割放疗的试验的 2426 例患者)。等剂量中度低分割放疗的中位随访时间为 5.4 年(四分位距4.6-7.2),剂量递增的中度低分割放疗的中位随访时间为 7.1年(5.7-8.4),未检测到无进展生存期方面的差异(HR=0.92,95% CI: 0.81-1.05;P=0.21和HR=0.94,95% CI: 0.82-1.09;P=0.43)。对于等剂量(优势比[OR]1.16,95% CI: 0.86-1.57;P=0.32)或剂量递增的中度低分割放疗(OR=1.20,95% CI: 0.95-1.51;P=0.13),均未发现 2 级或更高级别的泌尿生殖系统毒性效应的几率增加。剂量递增的中度低分割放疗出现2级或更高级别胃肠道毒性效应的几率显著更高(OR=1.48,95% CI: 1.14-1.92;P=0.0035),但等剂量中度低分割放疗则不然(OR=1.30,95% CI: 0.59-2.87;P=0.51)。未发现等剂量中度低分割放疗在泌尿生活质量下降(OR=1.03,95% CI: 0.51-2.09;P=0.93)或肠道生活质量下降(OR=0.76,95% CI: 0.40-1.43;P=0.39)的几率方面存在差异。剂量递增的中度低分割放疗与肠道生活质量下降的几率增加相关(OR=1.68,95% CI: 1.07-2.61;P=0.023),但未发现其会导致泌尿生活质量下降几率增加的证据(1.57,0.87-2.85;P=0.13)。
结论:与常规分割放疗相比,等剂量中度低分割放疗和剂量递增的中度低分割放疗疗效相似,但剂量递增的中度低分割放疗与医生评分以及患者报告的更高的肠道毒性相关。等剂量方案,例如 20 次分割给予 60 戈瑞,应该成为局限性前列腺癌的标准中度低分割放疗方案。
7.前列腺癌根治性前列腺切除术后前列腺特异性抗原持续存在与死亡风险
发表期刊:JAMA Oncol
重要性:前列腺癌(PC)根治性前列腺切除术(RP)后常规的 1.5 个月至 2.0 个月的时间间隔是否足以准确记录前列腺特异性抗原(PSA)持续存在的情况,目前尚无定论。
目的:评估根治性前列腺切除术后准确记录 PSA 持续水平所需的时间。
设计、场景和研究对象:本队列研究评估了以下两者之间是否存在显著的相互作用:(1)根治性前列腺切除术前 PSA 水平大于 20 ng/mL 与 20 ng/mL 及以下;(2)根治性前列腺切除术后PSA持续存在与 PSA 检测不到,对前列腺癌特异性死亡率(PCSM)风险和全因死亡率(ACM)风险的影响,并对已知的前列腺癌预后因素、根治性前列腺切除术时的年龄、根治性前列腺切除术的年份以及根治性前列腺切除术后放射治疗(RT)和雄激素剥夺治疗(ADT)的时间依赖性进行校正。还研究了 PSA 持续水平升高是否与更差的预后相关。纳入了 1992 年至 2020 年期间在 2 个学术中心接受根治性前列腺切除术治疗的 T1N0M0 至 T3N0M0 期前列腺癌患者。随访数据收集至 2023 年 11 月。数据分析时间为 2024 年 7 月至 2025 年 1 月。
暴露因素:根治性前列腺切除术。
主要结局和测量指标:全因死亡率和前列腺癌特异性死亡率风险的校正风险比(aHR)。
结果:在纳入发现队列的 30461 例患者中,中位(四分位距)年龄为64(59-68)岁;在纳入验证队列的 12837 例患者中,中位(四分位距)年龄为 59(54-64)岁。与 PSA 检测不到的患者相比,在 PSA 持续存在的患者中,根治性前列腺切除术前 PSA 水平大于 20 ng/mL 与 20 ng/mL 及以下相比,与全因死亡率风险降低显著相关(aHR=0.69;95% CI: 0.51-0.91;P=0.01;相互作用 P值<0.001)以及前列腺癌特异性死亡率风险降低显著相关(aHR=0.41;95% CI: 0.25-0.66;P<0.001;相互作用 P值=0.02)。在校正前列腺体积后,这一结果仍然成立,并且在验证队列中前列腺癌特异性死亡率风险方面得到了证实。这可能表明,与根治性前列腺切除术前 PSA 水平为 20 ng/mL 及以下的患者相比,根治性前列腺切除术前 PSA 水平大于 20 ng/mL 的患者中,有更高比例的患者如果在因推测 PSA 持续存在而开始根治性前列腺切除术后治疗之前有更多时间进行 PSA 评估,可能会达到 PSA 检测不到的水平。值得注意的是,根治性前列腺切除术前 PSA 水平大于 20 ng/mL 的患者(446 例患者中有 244 例[54.7%],中位[四分位距]时间为 2.68[1.51-4.40]个月)比根治性前列腺切除术前 PSA 水平为 20 ng/mL 及以下的患者(972 例患者中有 338例[34.8%],中位[四分位距]时间为 3.30[2.00-5.39]个月)更频繁且更早地接受根治性前列腺切除术后放疗联合雄激素剥夺治疗或雄激素剥夺治疗。这些治疗时间比观察到的患者达到 PSA 检测不到水平的时间更短(分别为中位[四分位距]2.96[1.84-3.29]个月和 3.37[2.35-4.09]个月)。PSA 持续水平升高与全因死亡率风险增加(aHR=1.14;95% CI: 1.04-1.24;P=0.004)和前列腺癌特异性死亡率风险增加(aHR=1.27;95% CI: 1.12-1.45;P<0.001)相关。
结论与意义:根治性前列腺切除术后至少 3 个月评估 PSA 水平可能会使过度治疗最小化,并且在本研究中,较高的 PSA 持续水平与较差的预后相关。
肾癌
8.局部晚期非转移性肾透明细胞癌的新辅助卡博替尼治疗:一项2期临床试验
发表期刊:Nat Cancer
背景:肾癌是全球发病率上升最快的疾病之一,在年轻患者和少数民族中尤为普遍。新辅助治疗策略最初是为了减小肿瘤大小,从而实现微创手术,如切除以前无法切除的肿瘤。虽然减少手术负担是这些治疗的最初目标,但越来越多的人认识到,在新辅助治疗中提供的一些治疗可以提高患者的长期生存率。卡博替尼是一种口服多激酶抑制剂,已获批用于治疗转移性肾细胞癌(RCC)。
方法:我们开展了一项2期、非随机、单臂临床试验(NCT04022343),对 17 例经活检证实的局部晚期、非转移性肾透明细胞癌患者,在手术切除前给予卡博替尼治疗 12 周。主要终点是第 12 周时的客观缓解率(完全缓解和部分缓解),次要终点包括安全性、耐受性、临床和手术结局以及生活质量。
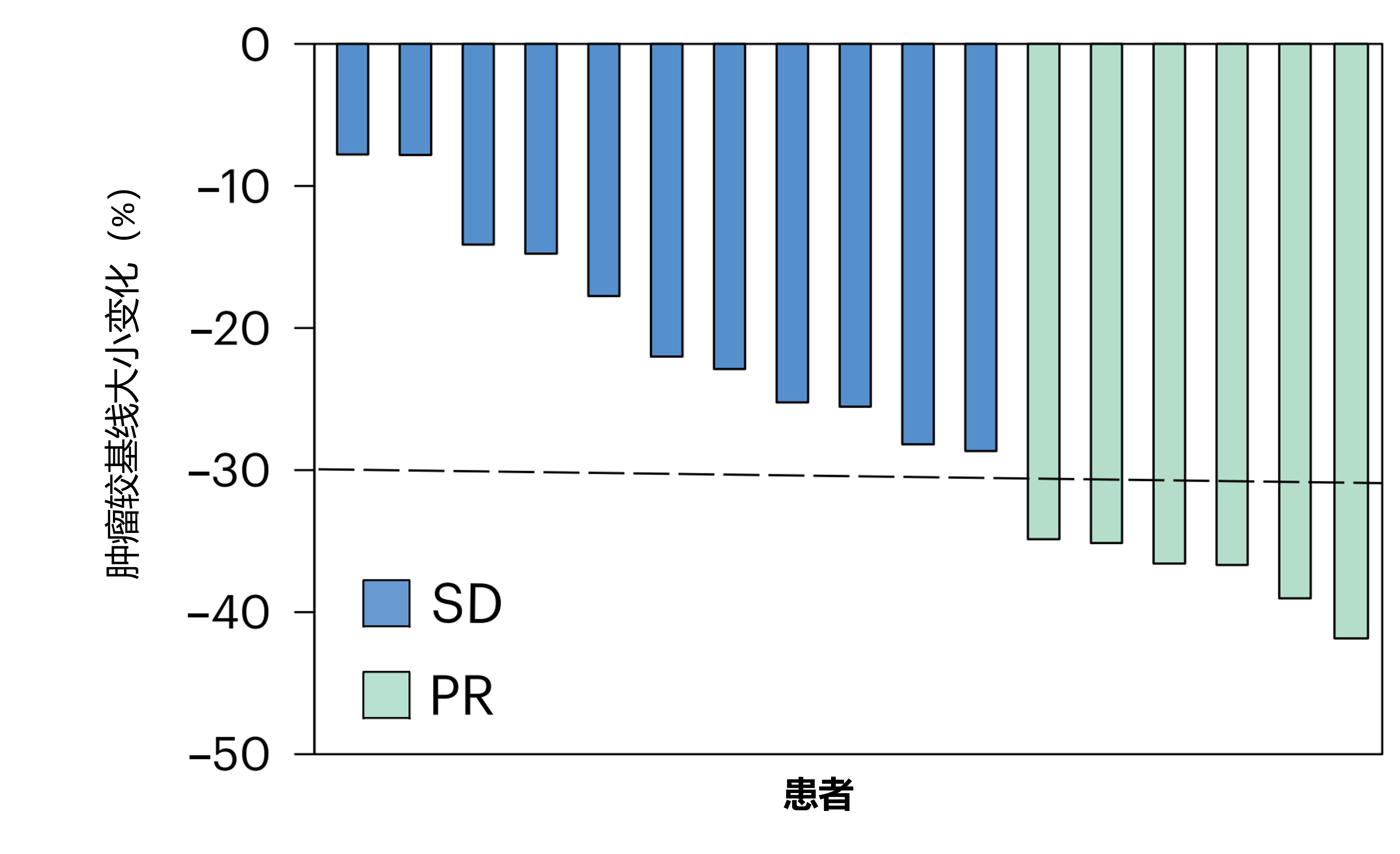
结果:6 例患者(35%)出现部分缓解,11 例患者(65%)病情稳定。最常见的不良反应为腹泻(n=12,70.6%)、厌食、疲劳和高血压(n=10,58.8%)、恶心和手足皮肤反应综合征(n=9,52.9%)。未发生与卡博替尼或手术相关的 4 级或 5 级治疗不良反应。1 年无病生存率和总生存率分别为 82.4%(95% CI: 54.7–93.9%)和 94.1%(95% CI: 65–99.1%)。卡博替尼治疗可激活血液中的 CD8+T 细胞,减少髓系细胞群体,并为 TCF1+干细胞样 CD8+T 细胞诱导免疫微环境。
结论:在局部晚期非转移性肾透明细胞癌患者的新辅助治疗中,卡博替尼具有临床活性且安全性良好。
图6.与基线相比肿瘤大小变化的百分比
9.晚期肾细胞癌患者使用贝组替凡对比依维莫司的健康相关生活质量(LITESPARK-005):来自一项随机、开放标签、3期试验的患者报告结局
发表期刊:Lancet Oncol
背景:基于 3 期 LITESPARK-005 试验的结果,首类缺氧诱导因子-2α(HIF-2α)抑制剂贝组替凡已获批用于曾接受过免疫检查点和抗血管生成治疗的晚期肾细胞癌患者。我们在此呈现 LITESPARK-005 试验中患者报告结局(PROs)的相关内容。
方法:LITESPARK-005 是一项开放标签、多中心、随机、活性药物对照的 3 期试验,在 6 个地区的 147 家医院和癌症中心开展。符合条件的受试者为年龄 18 岁及以上的晚期肾透明细胞癌患者,卡氏功能状态评分(Karnofsky Performance Status)为 70%及以上,根据实体瘤疗效评价标准(RECIST) 1.1 版有可测量的病灶,在接受抗 PD-1 或抗 PD-L1 免疫治疗以及一种血管内皮生长因子(VEGF)酪氨酸激酶抑制剂(序贯或联合使用)治疗期间或之后出现疾病进展,且此前接受的全身治疗方案不超过三线。符合条件的受试者通过交互式语音应答和网络应答系统以 1:1 的比例进行中心随机分组,分别接受口服贝组替凡 120 mg 每日一次或口服依维莫司 10 mg 每日一次。随机分组根据国际转移性肾细胞癌数据库联盟预后评分以及既往接受 VEGF 靶向或 VEGF 受体靶向治疗的次数进行分层。双主要结局为无进展生存期和总生存期,其结果此前已有报道。在本研究中,使用癌症治疗功能评估-肾癌症状指数:疾病相关症状量表(FKSI-DRS)和欧洲癌症研究与治疗组织生活质量核心问卷 30(EORTC QLQ-C30)对 LITESPARK-005 试验中预先设定的次要患者报告结局进行评估。患者报告结局分析人群包括所有接受至少一剂研究治疗且完成至少一次患者报告结局评估的受试者。使用受限纵向数据分析模型评估第 17 周时患者报告结局与基线相比的最小二乘均数变化。在患者报告结局分析人群中,还评估了由 EORTC QLQ-C30 评估的身体功能(预先设定)和角色功能(事后设定)的恶化时间。本试验仍在进行中,已停止招募,在 ClinicalTrials.gov 注册,注册号为 NCT04195750。
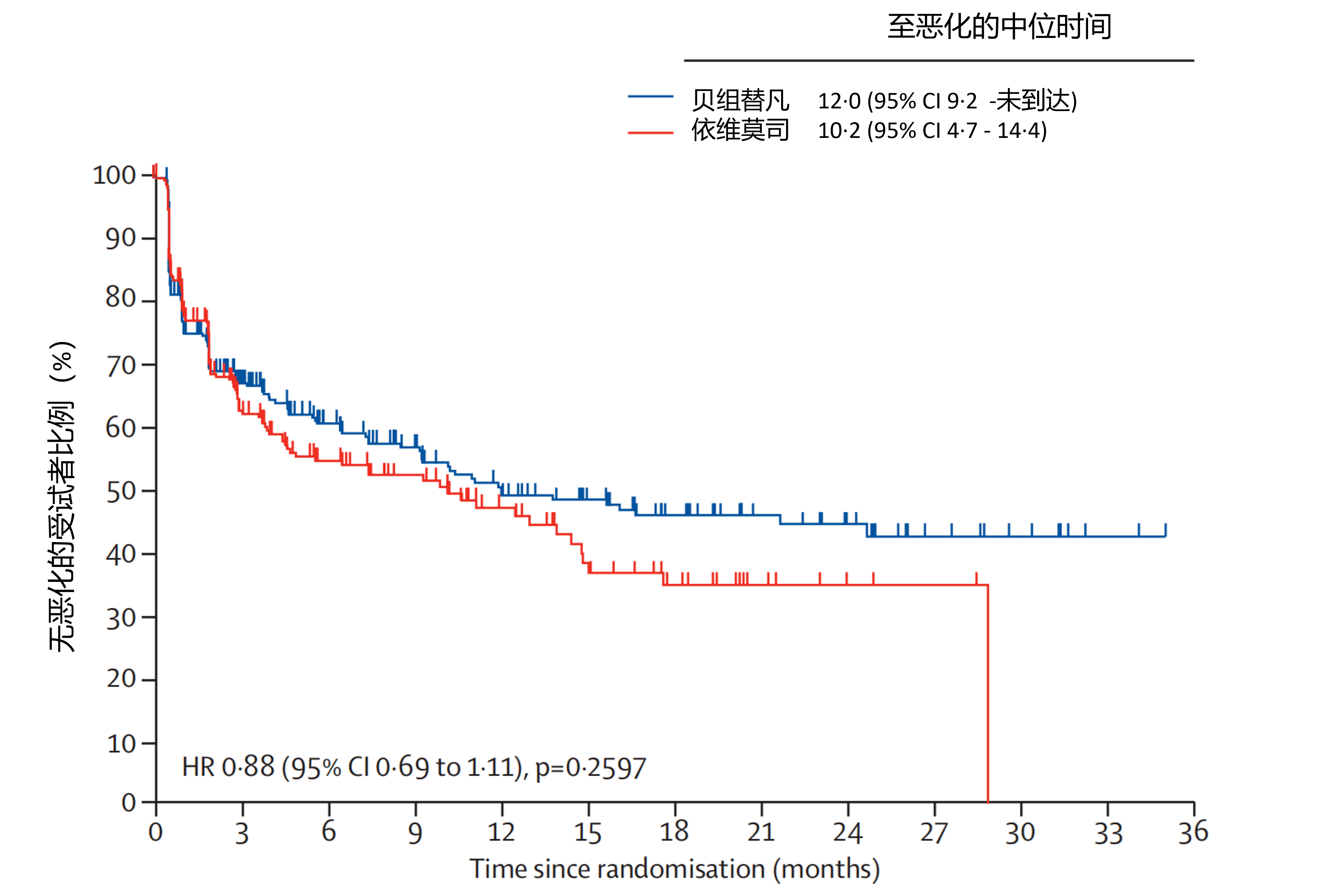
结果:在 2020 年 3 月 10 日至 2022 年 1 月 19 日期间,对 996 例受试者进行了筛查,746 例受试者被随机分配至贝组替凡组(n=374)或依维莫司组(n=372)。患者报告结局全分析集人群包括贝组替凡组的 366 例受试者和依维莫司组的 354 例受试者。从随机分组到数据库截止日期(2023 年 6 月 13 日)的中位时间为 25.7 个月(四分位距21.7-30.4)。在基线时,两组中 FKSI-DRS 和 QLQ-C30 的完成率均高于 94%,在第 17 周时均高于 55%。从基线到第 17 周,FKSI-DRS 评分的变化(两组最小二乘均数差值为1.5[95% CI: 0.7-2.2])以及 QLQ-C30 总体健康状况-生活质量(QOL)评分的变化(6.4[95% CI: 3.2-9.6])表明,与依维莫司导致情况恶化相比,贝组替凡组的情况保持稳定。两组之间从基线到第 17 周在 QLQ-C30 身体功能(最小二乘均数差值2.5[95% CI: −0.6-5.5])和 QLQ-C30 角色功能(4.2[95% CI: 0.1-8.4])子量表评分方面的变化相似。在 EORTC 身体功能方面,贝组替凡组和依维莫司组的恶化时间相似(贝组替凡组中位时间为 19.3 个月[95% CI: 11.1-未达到],依维莫司组为 13.8 个月[95% CI: 10.6-未达到];风险比0.93[95%置信区间0.72至1.20],见图 7),在角色功能方面也是如此(中位时间分别为 12.0 个月[95% CI: 9.2-未达到]对比 10.2 个月[95% CI: 4.7-14.4];HR=0.88[95% CI: 0.69-1.11],见图 8)。
结论:与依维莫司相比,贝组替凡用于晚期肾细胞癌与特定疾病症状的改善和生活质量的提高相关。结合 LITESPARK-005 试验的疗效和安全性数据,在这种情况下,贝组替凡可以在不影响患者生活质量的前提下提供临床获益。
图7.EORTC QLQ-C30身体功能子量表得分至恶化时间的Kaplan-Meier

图8.EORTC QLQ-C30角色功能子量表得分至恶化时间的Kaplan-Meier
10.伊匹木单抗/纳武利尤单抗对比标准治疗方案用于非透明细胞肾细胞癌的前瞻性随机II期试验——SUNNIFORECAST试验结果
发表期刊:Ann Oncol
背景:非透明细胞肾细胞癌(nccRCCs)是一组由 20 多种不同类型组成的异质性肿瘤,但很少被纳入大型随机试验中。无论是否联合免疫检查点抑制剂的酪氨酸激酶抑制剂被视为标准治疗方案(SOC),然而目前仍未确定最佳的治疗方法。我们设计了首个将伊匹木单抗/纳武利尤单抗与标准治疗方案进行对比的前瞻性随机试验。
患者与方法:我们将既往未接受过治疗的成年晚期或转移性非透明细胞肾细胞癌患者按 1:1 的比例随机分组,一组接受纳武利尤单抗 3 mg/kg 联合伊匹木单抗 1 mg/kg,每 3 周给药 1 次,共 4 剂,随后固定剂量的纳武利尤单抗,每 2 周 240 mg 或每 4 周 480 mg;另一组接受标准治疗方案。患者根据组织学类型和国际转移性肾细胞癌数据库联盟(IMDC)风险评分进行分层。必须进行中心病理检查。主要终点是 12 个月时的总生存率(OS),次要终点包括中位总生存期、缓解率、无进展生存期(PFS)、安全性和生活质量。
结果:总共有 157 例患者被分配接受伊匹木单抗/纳武利尤单抗治疗,152 例患者接受标准治疗方案。伊匹木单抗/纳武利尤单抗组的 12 个月生存率为78%(95% CI: 71-84%),而标准治疗方案组为 68%(95% CI: 60-75%,P=0.026)。如图 9,中位总生存期分别为 33.2 个月和 25.2 个月,P=0.163[HR=0.81(0.61-1.099)]。两组的无进展生存期相似[HR=0.99(0.77-1.28)]。客观缓解率(ORR)分别为 32.8%和 19.3%。乳头状和非乳头状肾细胞癌亚型之间在任何终点均未观察到显著差异。探索性分析显示,程序性死亡配体1(PD-L1)综合阳性评分(CPS)>1与显著的总生存期优势相关(HR=0.56[95% CI:0.37-0.86])。伊匹木单抗/纳武利尤单抗组中有 27 例患者(17%)因毒性而停药,标准治疗方案组中有 13 例患者(9%)因毒性而停药。
结论:伊匹木单抗/纳武利尤单抗在 12 个月这一关键节点显示出显著更长的总生存期,且毒性特征可接受。因此,我们的研究结果说明了与目前的标准治疗方案相比,伊匹木单抗/纳武利尤单抗对既往未接受过治疗的非透明细胞肾细胞癌患者中具有重要的临床获益。
图9.两组患者的总生存期

参考文献
- Mazzone E, Cannoletta D, Quarta L, et al. A Comprehensive Systematic Review and Meta-analysis of the Role of Prostate-specific Membrane Antigen Positron Emission Tomography for Prostate Cancer Diagnosis and Primary Staging before Definitive Treatment. Eur Urol [J]. 2025 Mar 27:S0302-2838(25)00155-1.
- Fay AP, Fizazi K, Matsubara N, et al. First-line talazoparib plus enzalutamide versus placebo plus enzalutamide in men with metastatic castration-resistant prostate cancer and homologous recombination repair gene alterations: patient-reported outcomes from the randomised, double-blind, placebo-controlled, phase 3 TALAPRO-2 trial [J]. Lancet Oncol. 2025 Apr;26(4):481-490.
- Fay AP, Fizazi K, Matsubara N, et al. First-line talazoparib plus enzalutamide versus placebo plus enzalutamide in men with metastatic castration-resistant prostate cancer and homologous recombination repair gene alterations: patient-reported outcomes from the randomised, double-blind, placebo-controlled, phase 3 TALAPRO-2 trial [J]. Lancet Oncol. 2025 Apr;26(4):481-490.
- Petrylak DP, Ratta R, Matsubara N, et al. Pembrolizumab Plus Docetaxel Versus Docetaxel for Previously Treated Metastatic Castration-Resistant Prostate Cancer: The Randomized, Double-Blind, Phase III KEYNOTE-921 Trial [J]. J Clin Oncol. 2025 Mar 5:JCO2401283.
- Aggarwal RR, Vuky J, VanderWeele D, et al. Phase I, First-in-Human Study of FOR46 (FG-3246), an Immune-Modulating Antibody-Drug Conjugate Targeting CD46, in Patients With Metastatic Castration-Resistant Prostate Cancer [J]. J Clin Oncol. 2025 Mar 26:JCO2401989.
- Kishan AU, Sun Y, Tree AC, et al. Hypofractionated radiotherapy for prostate cancer (HYDRA): an individual patient data meta-analysis of randomised trials in the MARCAP consortium [J]. Lancet Oncol. 2025 Apr;26(4):459-469.
- Tilki D, Chen MH, Wu J, et al. Persistent Prostate-Specific Antigen Following Radical Prostatectomy for Prostate Cancer and Mortality Risk [J]. JAMA Oncol. 2025 Mar 13:e250110.
- Bilen MA, Vo BT, Liu Y, et al. Neoadjuvant cabozantinib for locally advanced nonmetastatic clear cell renal cell carcinoma: a phase 2 trial [J]. Nat Cancer. 2025 Mar;6(3):432-444.
- Powles T, Choueiri TK, Albiges L, et al. Health-related quality of life with belzutifan versus everolimus for advanced renal cell carcinoma (LITESPARK-005): patient-reported outcomes from a randomised, open-label, phase 3 trial [J]. Lancet Oncol. 2025 Apr;26(4):491-502.
- Bergmann L, Albiges L, Ahrens M, et al. Interdisciplinary Renal Cell Carcinoma Working Group of the DKG (IAGN). Prospective Randomised Phase-II Trial of Ipilimumab/Nivolumab versus Standard of Care in non-clear cell Renal Cell Cancer - Results of the SUNNIFORECAST Trial [J]. Ann Oncol. 2025 Apr 1:S0923-7534(25)00124-3.



