
近几十年来,治疗方法的进步显著提高了激素受体阳性(HR+)乳腺癌患者的生存率。基于人群数据库的分析表明,与1990年代初确诊的患者相比,2015至2019年间患者总体死亡率显著降低22%,乳腺癌特异性死亡率下降27%。这种生存改善主要归功于内分泌治疗和靶向治疗策略的重大进展,尤其是细胞周期蛋白依赖性激酶4/6抑制剂(CDK4/6i)的应用,彻底改变了患者的治疗模式和预后状况。然而,目前缺乏可靠的生物标志物来识别潜在应答者及CDK抑制剂的早期耐药机制,这为晚期HR+乳腺癌领域实施精准医疗带来了重大挑战。
基于此,本文全面探讨HR+转移性乳腺癌(MBC)中CDK4/6i耐药机制的最新研究和前沿观点,通过整合现有耐药机制的相关证据,旨在为临床肿瘤学家提供可指导实践的深刻见解,以优化现有治疗策略 [1]。图1概括了CDK4/6i的主要耐药机制及相应的应对治疗药物方案。
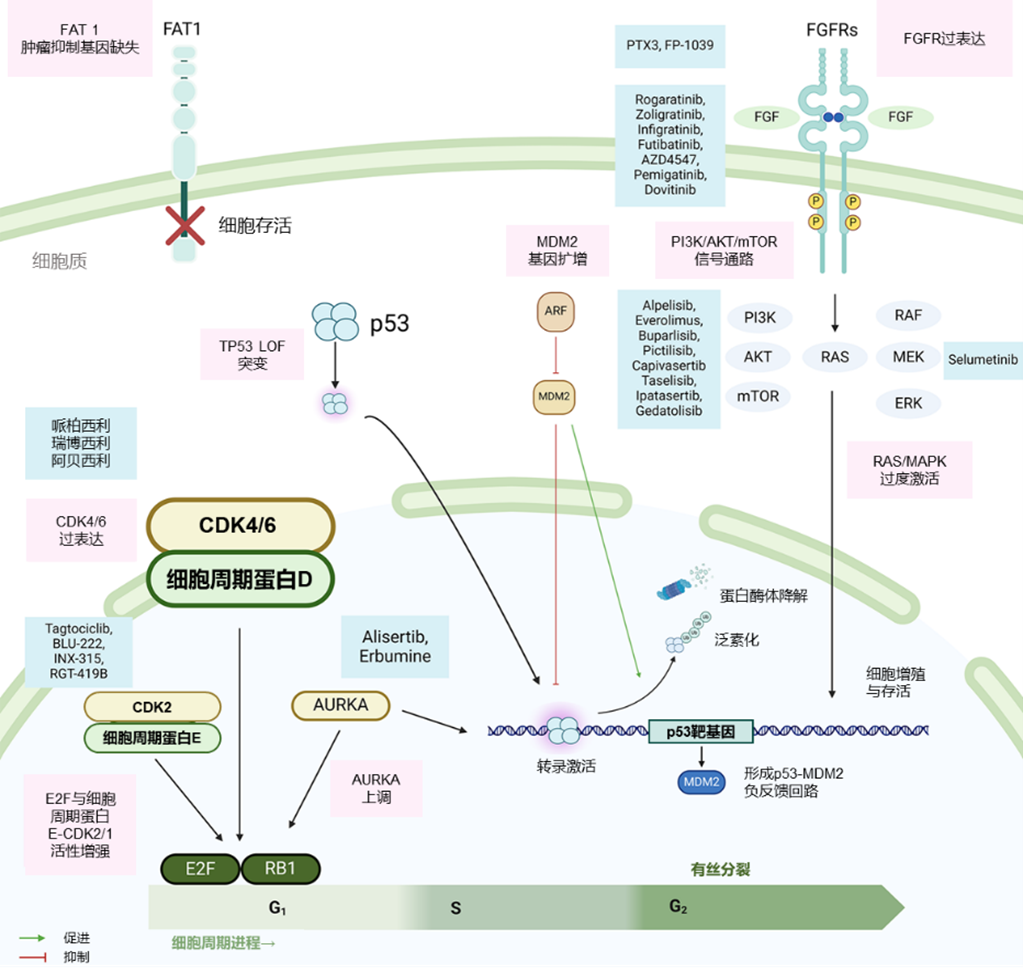
图1. CDK4/6i主要耐药机制及相应治疗药物方案
一、RB1分子改变
RB1作为关键抑癌基因,其编码的视网膜母细胞瘤蛋白(pRB)是细胞周期蛋白D-CDK4/6复合物的主要作用靶点。RB1基因的完整性对CDK4/6i的疗效至关重要,而该基因的改变可能提示原发或获得性耐药机制。多项研究证实,基线RB1突变在循环肿瘤DNA(ctDNA)分析中的检出率仅为0-5%。研究表明,携带RB1基因改变的肿瘤患者接受CDK4/6i治疗时,无进展生存期(PFS)获益会显著降低。
近期研究强调,特定RB1基因突变——尤其是"双打击"事件——在乳腺癌对CDK4/6i产生耐药方面具有重要作用。"双打击"是指RB1两个等位基因同时失活,可能原因是基因完全缺失、点突变或杂合性缺失(LOH),最终导致RB1功能完全丧失。这种遗传改变会加剧基因组不稳定性,增强肿瘤生存机制,从而促进恶性肿瘤的侵袭性。
在PALOMA系列研究中发现,少数患者出现新发RB1突变。这些基因突变在基线时并未检测到,表明RB1异常是在CDK4/6i治疗压力下获得性或选择性产生的。MONALEESA系列研究中ctDNA检测显示RB1失活突变或缺失的患者,从瑞博西利治疗中获得的生存获益较低,其中位PFS为3.8个月,而RB1野生型患者为9.2个月。
经CDK4/6i治疗后进展的RB1缺陷患者,或可从替代治疗策略中获益,如多腺苷二磷酸核糖聚合酶(PARP)抑制剂或靶向RB1下游效应分子的药物。因此,在乳腺癌治疗中,针对RB1的全面的基因分析(包括基因突变和"双打击"事件的动态监测),对于分层治疗策略和优化临床结局至关重要。
二、细胞周期调控机制失调
细胞周期调控机制失调是癌症的标志性特征,会导致细胞增殖失控。对于细胞周期蛋白D(cyclin D)或CDK4/6过表达/拷贝数扩增,同时伴有CDKN2A基因(CDK4/6活性的关键调控因子)失活的肿瘤,可能对CDK4/6i具有更高的敏感性。此外,下游基因改变(如RB1基因失活、E2F和E-CDK2复合物(cyclin E-CDK2)活性增强)可不依赖CDK4/6功能,直接促进细胞周期进程。尽管已有上述发现,但目前临床上唯一经过验证的CDK4/6i的生物标志物仍只有雌激素受体(ER)阳性。
HR+肿瘤通常与细胞周期蛋白 D1(cyclin D1)扩增和/或CDKN2A失活相关,表明这些肿瘤细胞的增殖主要受激素因素通过cyclin D1-CDK4/6活性驱动。另有充分证据表明,在 CDK4/6 活性被抑制的情况下,细胞会通过适应性激活 CDK2 来维持细胞周期进程。亦有研究显示,细胞周期蛋白E1基因(CCNE1)扩增或表达升高,可能会影响CDK4/6i疗效,这些研究结果使细胞周期蛋白 E1(cyclin E1)成为潜在的原发耐药生物标志物。然而,在缺乏充分证据证明 CCNE1 在不同人群、不同治疗场景中均具有预测价值的情况下,临床医生将其作为生物标志物应用时可能会面临阻碍。
三、成纤维细胞生长因子受体改变
在内分泌耐药的乳腺癌中,成纤维细胞生长因子受体(FGFR)通路的异常改变会导致肿瘤对 CDK 抑制剂产生耐药性。该通路通过成纤维细胞生长因子(FGFs)介导信号传导,调控细胞增殖与存活。在ET耐药的细胞中,FGFR1过表达会导致PI3K/AKT和RAS/MEK/ERK信号通路过度激活,这种异常激活有时甚至会产生配体非依赖性信号传导。
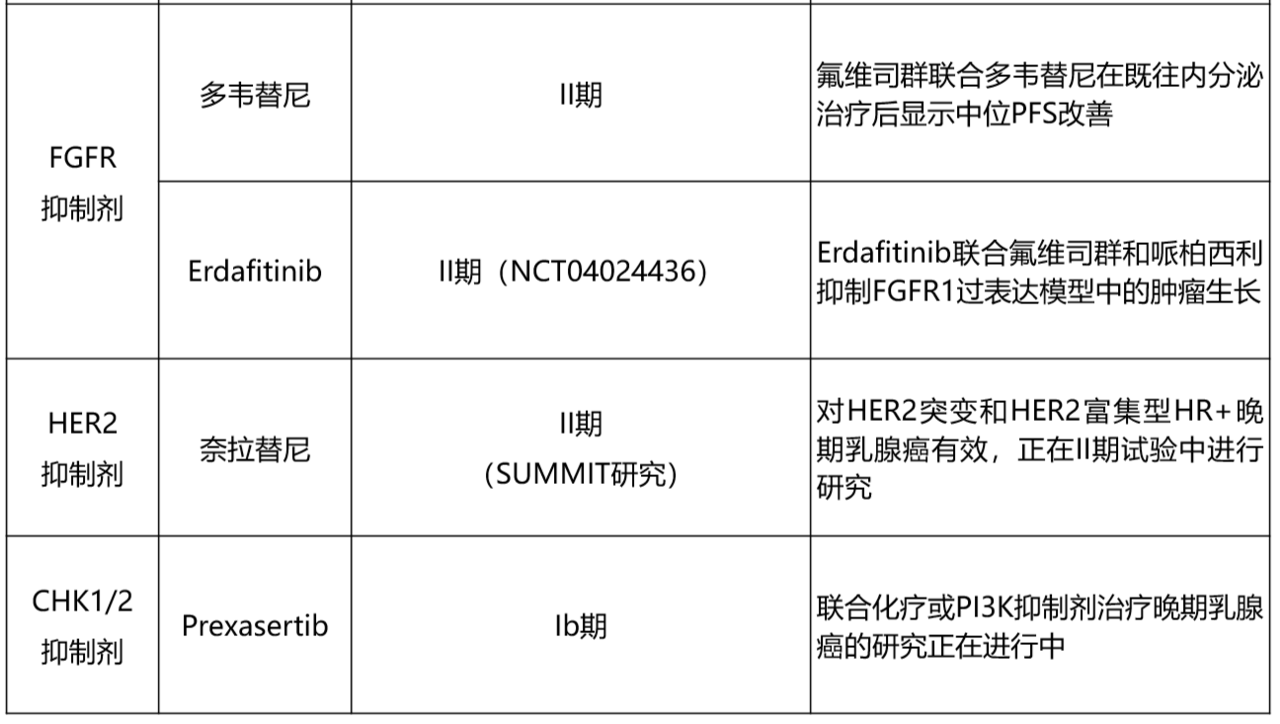
近期一项对哌柏西利联合ET治疗患者进行ctDNA的研究显示,34份样本中有 14 份检测出FGFR通路异常改变,主要以FGFR1扩增为主。在MONALEESA-2研究中,5%的患者出现FGFR1基因突变,这类患者的PFS较野生型患者缩短(10.6个月 vs 24.8个月,P=0.075)。然而,研究同时发现,无论患者FGFR1状态如何,瑞博西利的疗效未受影响。目前正在积极探索FGFR抑制剂联合CDK4/6i及ET的治疗策略。一项II期试验结果表明,氟维司群联合FGFR抑制剂多韦替尼较安慰剂组可延长中位PFS(10.9个月 vs 5.5个月)。
四、FAT1抑癌基因缺失、Hippo通路失活与CDK6过表达
抑癌基因(如FAT1)在调控细胞生长、分化和凋亡等细胞过程中发挥关键作用。导致FAT1基因功能缺失的突变可能会破坏其抑制细胞增殖的作用,导致癌细胞生长失控——尤其在HR+乳腺癌中。这类抑癌基因的改变会对治疗结局及患者预后产生显著影响。
既往研究显示,FAT1突变在HR+HER2乳腺癌中发生率为2-6%。FAT1基因缺失的患者对CDK4/6i呈现原发耐药性并且PFS获益受限。然而,由于FAT1基因突变率较低,因此对其进行常规肿瘤学筛查的临床价值优先。同时相关的检测成本与技术挑战可能使这类抑癌基因难以纳入标准临床实践。更重要的是,针对下游效应分子(如CDK6或YAP/TAZ)的治疗选择有限,突显了开发更有效治疗策略的迫切需求。
五、TP53通路改变(TP53功能缺失性突变、MDM2基因扩增)
TP53 突变在乳腺癌中的意义已引起广泛关注,主要因为TP53与治疗耐药性相关——尤其是在CDK4/6i治疗中。作为关键抑癌基因,TP53 在包括HR+乳腺癌在内的多种恶性肿瘤中常发生异常改变。研究表明,在对CDK4/6i长期疗效欠佳的患者中,TP53功能缺失性突变较为普遍。这些突变会破坏p53肿瘤蛋白的核心调控功能(包括细胞周期调控、DNA损伤应答和凋亡诱导),即便接受治疗干预,肿瘤细胞仍能维持存活与增殖能力。在HR+乳腺癌患者中,约30%~40%存在TP53突变,这迫切要求我们理解该患者群体临床特征,并为该群体探索靶向治疗方案。
TP53突变介导CDK4/6i耐药的机制十分复杂。在正常细胞中,p53蛋白允许CDK4/6i通过抑制pRB磷酸化来诱导细胞周期阻滞,从而阻止细胞周期进程。相反,在携带TP53突变的细胞中,这种调控通路被破坏,导致突变型p53蛋白促使细胞异常重新进入细胞周期并逃离静息状态(G0 期)——即便存在 CDK4/6 抑制剂,癌细胞仍能凭借这一机制持续增殖。
针对TP53介导的耐药性挑战,目前研究者们正在针对携带TP53突变的HR+乳腺癌患者开发替代治疗方案。其中一种有前景的策略是选择性抑制细胞周期蛋白依赖性激酶 2(CDK2),该策略已展现出克服p53功能缺失效应并恢复细胞周期调控的潜力。通过靶向CDK2,可能增强TP53 突变肿瘤对治疗的应答,进而改善临床结局。此外,将基因谱分析整合到临床实践中,有助于识别可能从此类靶向治疗中获益的患者,为实现更精准的个体化治疗策略奠定基础。
六、Aurora激酶上调
Aurora激酶A(AURKA)已成为介导HR+ MBC对CDK4/6i产生耐药性的关键因子。AURKA作为一种丝氨酸/苏氨酸激酶,对细胞周期调控至关重要,尤其在有丝分裂阶段发挥核心作用。在对CDK4/6抑制剂治疗耐药的患者群体中,AURKA的异常改变(如基因扩增和过表达)普遍存在。研究表明,约26.8%的耐药患者肿瘤样本中存在AURKA扩增,而治疗应答者的样本中基本不存在该异常改变。这一发现表明AURKA的异常改变可能通过促进耐药,使癌细胞逃避CDK4/6i的生长抑制效应,进而导致肿瘤进展并降低患者生存结局。在接受CDK4/6i治疗的患者中,AURKA的异常改变与不良临床结局相关。具体而言,携带AURKA扩增的患者较无AURKA扩增的患者PFS通常更短。更重要的是,AURKA通过参与RAS/ERK等增强细胞增殖和存活的信号通路,在介导耐药过程中发挥关键作用。AURKA过表达导致的这些通路激活会加速细胞周期进程并增强抗凋亡能力,最终导致治疗失败。
鉴于AURKA在耐药机制中的关键作用,AURKA抑制剂已成为应对这一挑战的潜在治疗策略。选择性AURKA抑制剂LY3295668在一项I期单药安全性研究中展现出疗效:在12例局部晚期实体瘤患者中,疾病控制率达到69%。另有一项II期试验纳入91例ET耐药且CDK4/6i耐药的绝经后MBC女性患者,评估了alisertib单药治疗与alisertib联合氟维司群治疗的效果。结果显示,两组治疗方案的缓解率相似(均约为 20%),alisertib单药显示出抗肿瘤活性,但联合用药未改善治疗结局。
七、PI3K/AKT/mTOR信号通路过度激活
PI3K/AKT/mTOR信号通路是调控细胞生长、代谢及存活的重要细胞网络。在HR+乳腺癌中,该通路的过度激活已成为导致肿瘤对标准治疗(尤其是CDK4/6i)产生耐药的重要因素。在HR+/HER2− MBC中,约50%患者存在该通路的异常改变,包括PI3KCA基因突变及抑癌基因PTEN的功能缺失。这类异常会增强cyclin D的表达与活性,最终降低 CDK4/6i的疗效。临床研究一致表明,PI3K/AKT/mTOR通路异常与HR+ MBC患者较差的生存结局相关——携带PIK3CA突变或PTEN缺失的患者在接受ET联合CDK4/6i治疗后,PFS和总生存期(OS)均显著缩短。研究还发现,内分泌治疗耐药的患者在启动 CDK4/6 抑制剂治疗前,其PI3K/AKT/mTOR通路的激活水平已显著升高,这进一步凸显了该通路在耐药机制中的作用。
然而,在此背景下,区分预后生物标志物与预测生物标志物至关重要:PI3K/AKT/mTOR通路异常属于预后生物标志物,可用于评估疾病进展趋势及患者总体结局,但无法作为指导CDK4/6i治疗决策的预测生物标志物。因此,尽管这类通路异常提示患者预后较差,却无法帮助肿瘤科医生判断CDK4/6i对特定患者的可能疗效。
针对PI3K/AKT/mTOR通路的特异性抑制剂的出现,显著改变了HR+ MBC的治疗格局,尤其为既往ET进展的患者提供了新选择。现有数据显示,尽管PI3K/AKT/mTOR抑制剂联合CDK4/6i在HR+ MBC中具有明确疗效,但不同PI3K/AKT/mTOR抑制剂在疗效和耐受性方面不可相互替代——大多数抑制剂无论是单药还是联合方案,均被证明难以实际应用。因此,安全性、生活质量和患者报告结局(而非单纯疗效)应成为评估这些靶向药物临床研究的核心要素。
八、RAS/MAPK信号通路过度激活
丝裂原活化蛋白激酶(MAPK)通路的激活(主要通过NF1、KRAS、BRAF、MAP2K1等组分实现)在乳腺癌内分泌耐药中发挥重要作用。这些基因的突变在早期乳腺癌中较为罕见,而在MBC中发生率更高——其中NF1基因(可负向调控RAS活性)的突变尤为突出。NF1功能缺失会导致RAS过度激活,进而同时介导肿瘤对内分泌治疗的原发耐药与获得性耐药。
ERK通路(MAPK通路的核心分支)与ER信号通路存在复杂的交互作用:雌激素的非基因组效应会快速激活RAS及其下游通路。这种激活不仅会增强雌激素对肿瘤细胞的增殖与存活促进作用,还会导致受体酪氨酸激酶(RTKs)过表达,而RTKs过表达会进一步加剧内分泌耐药。此外,RTKs与ER自身均可激活PI3K通路和ERK通路,这两种通路的激活会导致ER共调节因子发生磷酸化,进而增强ER活性,最终推动耐药机制形成。有证据表明,在接受他莫昔芬等内分泌治疗的患者中,RAS、RAF、ERK 的磷酸化水平与不良结局相关,这凸显了MAPK通路激活对临床预后的影响。
尽管在临床前研究中,使用ERK通路抑制剂克服内分泌耐药的策略展现出潜力,但临床研究结果并未一致支持这类方案的有效性。对接受芳香化酶抑制剂(AI)治疗期间疾病进展患者的ctDNA分析发现,部分患者存在RAS突变,这一结果进一步揭示了与MAPK通路激活相关的耐药机制的复杂性。
九、"非管腔型"内在亚型
在临床前与临床场景中,管腔型乳腺癌对CDK4/6i均表现出敏感性。这一观察结果得到了来自HR+乳腺癌、三阴性乳腺癌(TNBC)及人表皮生长因子受体2阳性(HER2+)乳腺癌的临床前研究数据支持。然而,CDK4/6i在非管腔型HR+/HER2−乳腺癌中的疗效仍不明确。
有相当比例(5%~20%)的HR+肿瘤可能表现出非管腔型特征,这可能意味着肿瘤对ET的敏感性降低,而对化疗的应应性增加,从而可能影响整体预后。此外,肿瘤进展还与表型转变相关,即向侵袭性更强、不依赖ER的非管腔型表型发展。
针对MONALEESA III期临床试验中针对内在亚型生物标志物与治疗反应的回顾性分析显示,大多数乳腺癌亚型均从“ET联合CDK4/6i”方案中显著获益。值得注意的是,该队列中HR+基底样型肿瘤(共 309 例,占2.6%)表现出cyclin E1和表皮生长因子受体(EGFR)高表达,同时管腔相关基因低表达,表型与TNBC相似。另一项有趣的发现是:在 HER2 富集亚型的 HR+/HER2−患者中,仅接受ET的患者预后较差;而在“ET联合CDK4/6i”方案中,这类患者获得的获益更为显著,且在再次活检时,相当比例初始为HER2富集亚型的肿瘤转变为管腔A型亚型。
目前,CDK4/6i对非管腔型内在亚型患者的疗效获益尚需进一步研究。一项国际II期临床试验(PONTIAC; NCT06486883)已启动,旨在评估德曲妥珠单抗(T-DXd)对比基于CDK4/6i的内分泌治疗,作为经PAM50归类为非管腔型的HR+、HER2低表达/超低表达MBC患者一线治疗方案的安全性与有效性。值得注意的是,一项荟萃分析发现,在接受后线或一线CDK4/6i联合ET的患者中,HER2低表达状态与客观缓解率(ORR)或OS均无显著相关性。对PALOMA-2和PALOMA-3试验的二次分析表明,CDK4/6i联合ET可改善低表达阳性患者PFS,而HER2零表达患者的获益主要见于既往ET进展的人群。另一项前瞻性研究也未能证实HER2低表达状态可作为HR+ MBC患者使用CDK4/6i联合ET的预测性生物标志物。
十、致病性胚系突变
乳腺癌中经常观察到胚系与体细胞致癌突变的共存现象,然而其联合生物学及临床意义尚未得到充分评估。最新研究表明,致病性胚系突变——特别是胚系BRCA1/2(gBRCA1/2)突变——可能通过双重机制影响CDK4/6i的疗效。一方面,BRCA2突变肿瘤通常表现出更高的基因组不稳定性,因而可能使其对CDK4/6i和DNA损伤剂联合治疗方案更为敏感;另一方面,这些突变通过重构细胞周期调控机制,可能引发适应性耐药,从而降低CDK4/6i单药疗效。
为评估胚系-体细胞相互作用对临床结局的影响,Safonov等人开发了整合临床-基因组学分析流程,对超过4500例乳腺癌患者的基因组进行了分析:结果发现gBRCA2相关肿瘤富含RB1功能缺失突变,且对一线CDK4/6i治疗反应不佳。在这些肿瘤中,gBRCA2相关的同源重组缺陷(HRD)和基线RB1杂合性缺失(LOH)状态,会在CDK4/6i的选择压力下促进RB1功能缺失突变,最终导致耐药。这些发现提出了一种替代治疗策略:对于胚系BRCA基因突变的乳腺癌患者,可在CDK4/6i治疗前,先使用PARP抑制剂靶向HRD缺陷,以此减轻RB1缺失导致的有害进展轨迹并抑制CDK4/6i耐药性的产生。更广泛地说,这些重要发现揭示了胚系-体细胞驱动的基因组构型如何影响系统治疗反应,并可通过生物标志物导向的临床实践策略进行治疗优化。
越来越多的真实世界证据表明,携带DNA损伤修复相关基因(如BRCA1、BRCA2和PALB2)致病性胚系变异的患者,接受CDK4/6i治疗效果欠佳。这些发现得到进一步支持:MonarchE试验的探索性分析中,3.5%的患者携带gBRCA1/2突变。其中阿贝西利联合ET组20例患者中仅1例(5%)出现无浸润性疾病生存期(iDFS)事件,而单纯ET组21例患者中有9例(42.8%)发生iDFS事件。
总结
尽管HR+ MBC治疗取得重大进展,但CDK4/6i耐药仍是一项严峻挑战,不仅影响治疗的长期疗效,更会阻碍预后的改善。耐药机制的复杂性(如RB1和PIK3CA基因突变、CDK2与cyclin E失调等)凸显了开发靶向生物标志物策略以预测治疗结局的重要性。此外,部分患者可能对CDK4/6i联合ET的一线方案产生原发性耐药导致生存期缩短,而长期接受该治疗方案也可能引发获得性耐药。未来研究必须优先关注以下核心领域:(1)深入阐明CDK4/6i的作用机制;(2)明确CDK4/6i与现有各类ET方案的协同作用;(3)优化CDK4/6i联合ET进展后的挽救治疗方案;(4)探究CDK4/6i联合ET的耐药机制。最后,将ctDNA检测整合到实时动态监测中,有望实现对获得性耐药的早期识别,并为个体化治疗策略的制定提供支持。通过聚焦这些关键领域,有望在HR+ MBC这一充满挑战的领域提升治疗效果并改善患者结局。
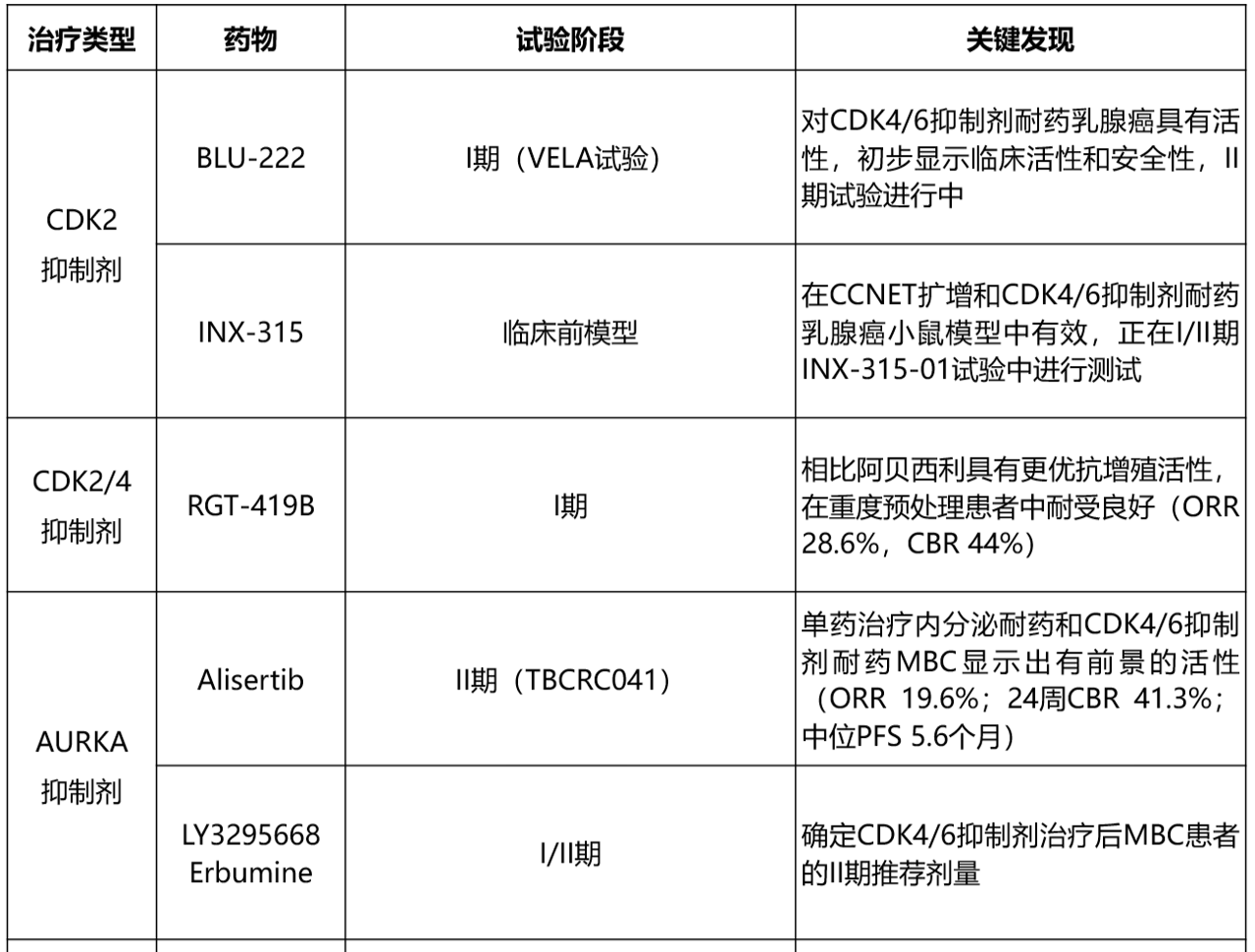
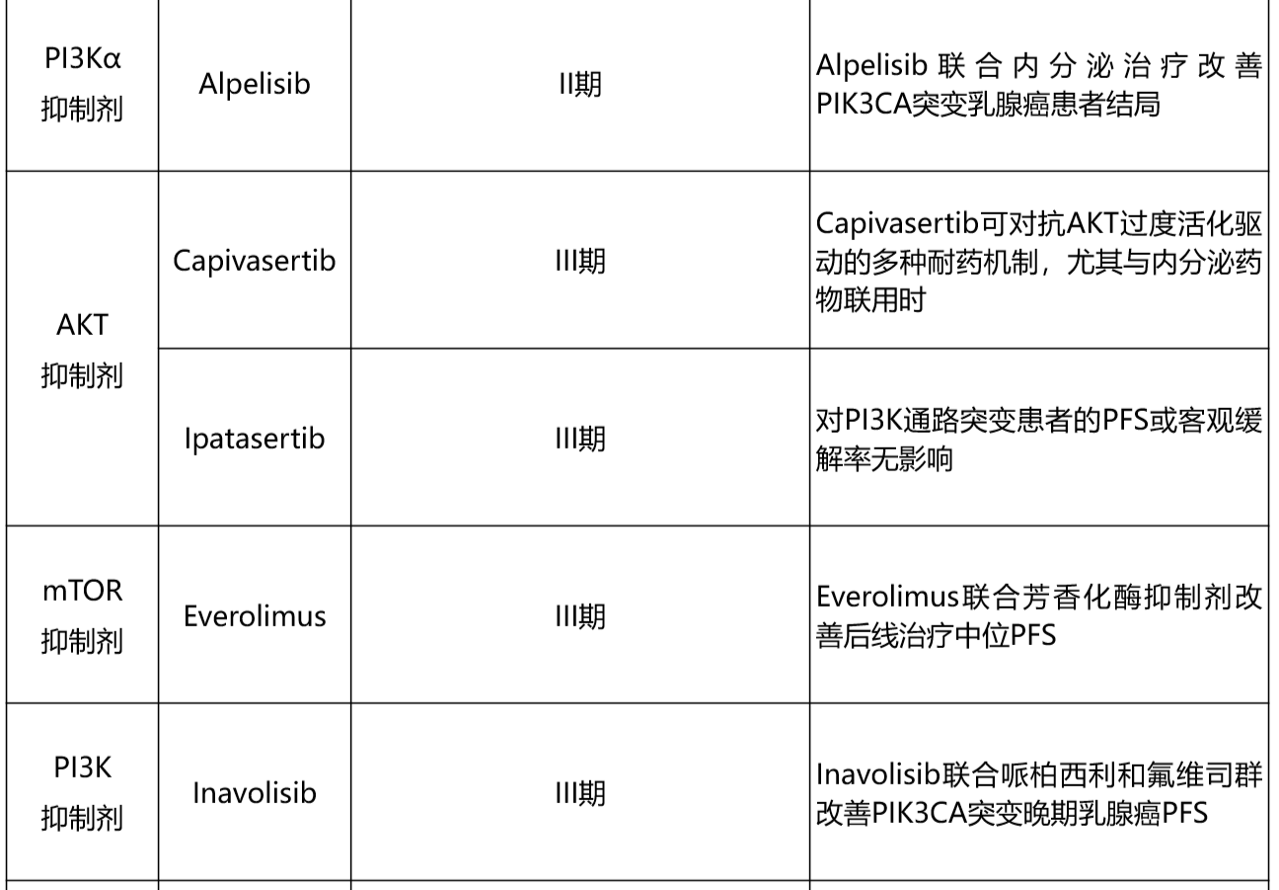
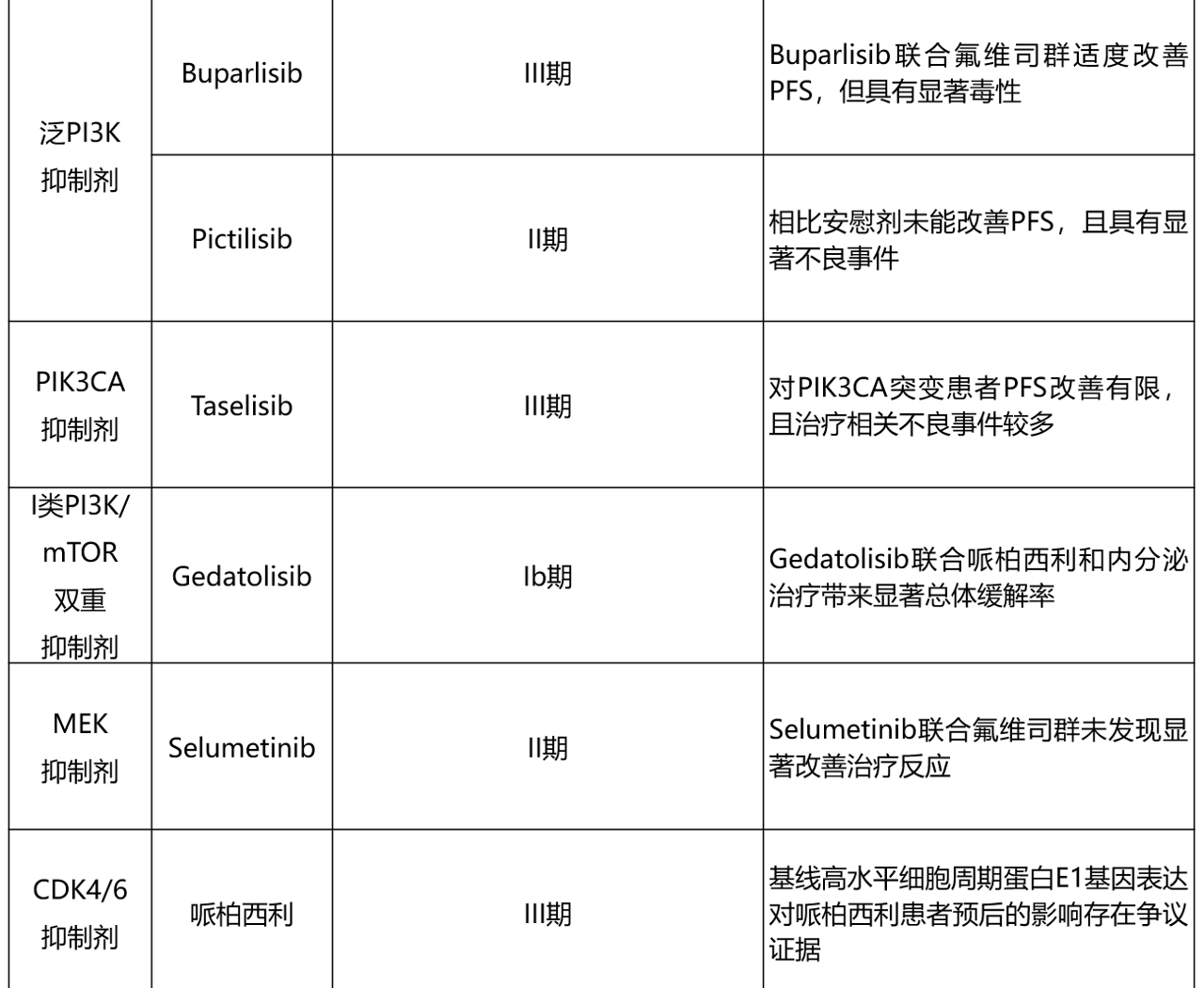
表1总结了HR+乳腺癌领域报告CDK4/6i耐药相关生物标志物发现的关键临床试验。
表1. 已发表的CDK4/6抑制剂耐药潜在生物标志物关键临床试验
参考文献
- da Silva JL, Oliveira LJC, de Resende CAA, et al. Understanding and overcoming CDK4/6 inhibitor resistance in HR+/HER2- metastatic breast cancer: clinical and molecular perspectives[J]. Ther Adv Med Oncol. 2025 Jul 10;17:17588359251353623.

免责声明:此资料应仅在其原始链接位置观看。对于经其他途径查看到的内容,辉瑞不承担责任。请扫描文档旁边的二维码,以获得说明书信息。



